
Apoptosis of pancreatic β-cells in type 1 diabetes: The role of Caspase-3 in β-cell apoptosis
Tatsuo Tomita1*
1Departments of Integrative Bioscience and Pathology, Oregon Health and Science University 611 SW Campus Drive, Portland, Oregon 97239-3097, USA
Abstract
Type 1 diabetes (T1D) results from autoimmune destruction of pancreatic β-cells after asymptomatic period over years. Insulitis activates antigen presenting cells, which trigger activating DC4+ helper-T cells, releasing cytokines. Cytokines activate CD8+ cytotoxic –T cells and lead to β-cell destruction. Apoptosis pathway consists of extrinsic and intrinsic pathway. Extrinsic pathway includes Fas pathway to CD4+-CD8+ interaction whereas intrinsic pathway includes mitochondria-driven pathway at balance between anti-apoptotic Bcl-2 and Bcl-xL and pro-apoptotic proteins. Activated cleaved caspase-3 (ACC) is the converging point between extrinsic and intrinsic pathway. ACC may be used as a marker for β-cell apoptosis: inT1DM islets weakly insulin-positive β-cells are present with increased ACC positive β-cells whereas β-cell absent islets, which exclusively consist of α-cells and PP-cells with decreased ACC-cells, represent the end-stage of regenerating β-cells. Weakly insulin-positive β-cells could be rejuvenated to supply endogenous insulin. Apoptosis takes place only when pro-apoptotic protein exceeds anti-apoptotic proteins. Since concordance rate of T1D in the identical twins is about 50%, environmental factors are involved in development of T1D, opening a door to means to prevent autoimmune β-cell destruction for therapeutic application by transfecting β-cells with Casp3-/- genes or pan-caspase inhibitor therapy. Prudent glucose control prevents ongoing hyperglycemia-induced β-cell apoptosis.
Introduction
Type 1 diabetes (T1D) results from severe insulin deficiency by loss of insulin-producing β-islet cells and it develops mostly in young and accounts for 5 to 10% of the diabetic subjects<su/s1,2up>1,2. T1DM develops as the consequence of progressive β-cell destruction by autoimmune processes after an asymptomatic period over several years2. MHC class II are expressed at the surface of antigen presenting cells (APC) (e.g. dendritic cells, macrophages and B-lymphocytes) including DR, DQ and DP and some specific combination of alleles for DQA1 and DQB1 genes and DRB1 genes (DRB1*03 and DRB1*04) significantly increase the development of T1D2-5. The earliest sign of autoimmunity against β-cells are often detectable months or years of clinical T1DM and the most common autoantibodies in pre-diabetic subjects are directly against glutamic acid decarboxylase (GAD65), tyrosine phosphatase-like protein (IA-2) and insulin6. Up to 90 % of newly diagnosed T1DM subjects have autoantibodies to one or more of these antigens6 and autoimmunity detection rates increase to 98% when combining the detection of these three antibodies plus antibodies against the newly discovered β-cell autoantigen ZnT87,8.
In identical twins, the concordance rate is below 50% for T1D as compared to a higher concordance rate for type 2 diabetes (T2D), suggesting that environmental or non-genetic factors contribute for T1D as also in the case for T2D, the latter has the concordance rate of about 90% in the identical twins9. Epidemiological data suggest that autoimmune process is triggered early in life, which may indicate that the pool of self-reactive naive T-cells stay in the control of the immune system for several years with some antibody positive subjects never develop insulitis nor proceed to overt T1D10. The environmental factors include viral infection, including that of Coxsackievirus B (CVB), rubella, mumps, rotavirus and cytomegalovirus11, and toxins, dietary factors during infancy, vaccination and others12,13. With immunocytochemical staining of 29 cases of pancreata from the T1D subjects who died within 18 months of diagnosis, Wilcox et al reported the following findings: CD 8+ cytotoxic cells were most abundant population in insulitis; CD 68+ macrophages were also present in insulitis whereas CD 4+ helper cells were present in the islet infiltrates, but were less numerous than CD 8+ or CD 68+ cells whereas CD 20+ cells increased during late insulitis since CD 20+ cells were recruited late during insulitis14. β-cell damage and destruction are mediated through CD 8+ and CD 4+ but natural killer cells (NK cell) do not appear to be required for β-cell death14. Insulitis is defined by at least 15 CD 45+ cells/islet present in three or more islets15 and CD 8+ cells are predominant15. Only 10%--30% of islets show insulitis at any time, even at the time of T1D diagnosis16, and there is different stage of islet destruction within the same pancreas. The first detection of autoantibodies peaks at one to three years of age in children who develop early T1DM and the second incidence peak is seen around puberty and shows more heterogenous autoantibody profile than in early forms of T1D17. T1D follows a slow-progressing autoimmune process before presenting typical clinical symptoms through a cascade of complicated immunological sequences as reported by Eizirik and his associates as follows3:
Autoimmune destruction of pancreatic β-cells
A) After the early stages of insulitis, activated local APCs recruit and activate CD4+ helper T-cells via migration to the pancreatic lymph nodes to present β-cell antigen and release of chemokines/cytokines3 (Fig. 1A).
B) CD4+ helper T-cells in turn stimulates APC secretion of cytokines and nitric oxide (Fig. 1B).
C) Cytokines induce secretion of chemokines by endothelial cells which enhance to recruit immune cells into the islets and activate CD8+ cytotoxic T-cells together with cytokines (Fig.1C).
D) β-cells also secrete chemokines in response to viral infection or cytokines, enhancing recruitment and activation of immune cells. Activated CD8+ cells in turn induce β-cell apoptosis (Fig. 1D).
E) Fas pathway: Fas (CD 95), a member of TNF superfamily is activated via binding of Fas L (CD 178) and Fas and Fas L are detected respectively at the surface of β-cells and T-cells infiltrating islets3. Once activated, Fas trimerizes and recruits the Fas-associated death domain (FADD), which recruits pro-caspase-8 leading to its activation by autocleavage. Activated caspase-8 subsequently cleaves the effector caspase-3 and/or activates the BH3 protein Bid (Fig. 1E).
F) The perforin/granzyme system: Perforin and granzyme are contained in granules inside the CD8+ T-cells. Perforin is involved in pore formation across the membranes via Ca2+ dependent mechanisms. The pore enables the entry of serine protease granzyme inside the cell, causing the cleavage of and activation of several targets, such as effector caspase-3 and the BH3 protein Bid.(Fig. 1F)
G) Interleukin-1β (Il-1β) activates NFkB and the kinases PKC, p38 and JNK: IL-1β binding to its receptor induces the formation of multiproteic complex at its cytoplasm domain including IL-IRAcP, Tollip, MyD88 and IRAK family, namely IRAK-1 and IRAK-4 (Fig.1G).
H) Tumor necrosis factor α (TNF-α) activates caspase-8, NF-kβ and the MAPK p38 and JNK3. Activation of TNF receptor 1 (TNF-R1) upon TNF-α binding leads to its trimerization and recruitment of adaptor protein TNF-associated death domain protein (TRADD), which in turn recruits TRAF-2 and serine threonine kinase Rip. TRAF-2 activates NF-kB and MAPK pathways. TNF-α phosphorylates and activates p38 and JNK. Caspase-8 activates effector caspase-3 in the same way in the Fas pathway.(Fig. 1H)
I) Interferon ϒ (IFN-ϒ) activates Stat-1 and the kinase ERK: IFN-ϒ binding to its receptor induces its oligomerization and cytoplasmic recruitment of two members of Janus kinase (Jak) family, Jak-1 and Jak-23, which recruit Stat-1 and trigger its activation by phosphorylation. Stat-1 then homodimerizes and migrates to nucleus where it regulates expression containing ϒ-activated sequence (GAS) elements such as caspases, Fas and iNOS (Fig. 1)3.
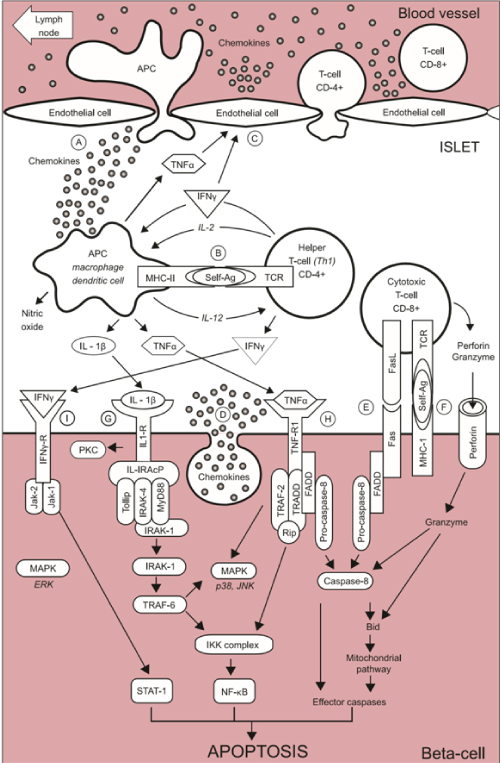
Figure 1. Schematic representation of the auto-immune attack to the beta-cells inT1D.
(A) At the early stage of insulitis, activated local antigen presenting cells (APC) recruit and activate CD4+ helper T-cells via migration to the pancreatic lymph node, presentation of beta-cells antigens and release of chemokines/cytokines. (B) CD4+ helper T-cells, in turn, stimulates APC secretion of cytokines and nitric oxide. (C) Cytokines induce the secretion of chemokines by endothelial cells which enhance the recruitment of immune cells into the islets and, together with cytokines, activate CD8+ cytotoxic T-cells. (D) The beta-cell themselves also secrete chemokines in response to viral infection or cytokines, further enhancing the recruitment and activation immune cells. Activated CD8+ cytotoxic T-cells, in turn, induce beta-cell apoptosis via (E) the Fas pathway and (F) the granzyme /perforin system. Cytokines also bind to receptors at the surface of beta-cells: (G) Interleukin-1β (IL-1β) activates NF-κB and the kinase d PKC, p38 and JNK (H)Tumor necrosis factor α (TNF α) activates caspase-8 and the MAPK p38 and JNK (I) Interferon ϒ (IFNϒ) activates Stat-1 and the kinase ERK. From reference 3. From reference 3. Pirot,P, Cardozo, AK, Eizirk, DL, Arq Bras Endocrine Meatv 2008;52(2):156-165.
In this theory, activated caspase-8 cleaves and activates effector caspase-3 and BH3 protein Bid in the E) Fas pathway and H) TNDα pathway and in part in I) IFNY pathway3.
Extrinsic and intrinsic apoptosis pathway
There are two main apoptosis pathways, the extrinsic (receptor-mediated) and the intrinsic (mitochondrial-driven) pathway. Above described theory refers mostly extrinsic pathway and is not distinctively clarified into extrinsic or intrinsic pathway, respectively and each pathway will be described below despite some redundancy between the above and below description on apoptosis (Fig. 2)19.
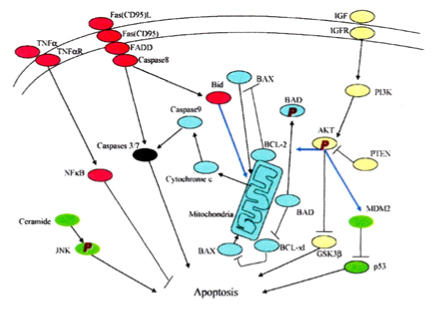
Figure 2. There are the extrinsic (receptor-mediated, red) and intrinsic (mitochondria-driven, blue) apoptosis pathways as opposed to the survival proteins such as the P13/Akt signaling circuity (yellow). From reference 14. Lee, SC, Pervaiz, S. Int J Biochem 2007; 39(3):497-504.
It is the extrinsic pathway that is activated upon ligation of the cells surface death receptors (s), which in turn activate(s) a downstream effector mechanism orchestrated by the caspase family of cysteine proteases (Fig. 2)19. The prototype example of death signaling via the extrinsic pathway is the Fas death receptor (FDR), which instigates assembly of the death-inducing signaling complex (DISC), a multi-protein complex comprising the cytoplasmic aspects of the Fas receptor, the adaptor protein FADD and preocaspase-8 (Fig. 2)19. Caspase-3 is the converging point of the apoptotic pathway (Fig. 2)19 and its peptide inhibitors have been shown to prevent islet apoptosis and improve islet graft function21. Apoptosis induced by ligation of cell surface receptors like Fas L or TNF receptors, ”death receptors” represents a pathway controlled by caspases22,23. Ligand binding of the receptor causes assembly of series of proteins of the death-inducing signaling complex, which then activates apical caspase, procaspase-819,24. The resulting events proceed in cascade that caspase-8 induces activation of caspase-321. Apoptosis manifests in two major execution programs, downstream of the death signal, the caspase pathway25 and upstream of irreversible cellular damage reside the Bcl family members, which are proteins with both anti-apoptotic and pro-apoptotic properties, playing a pivotal role in the life and death of cells (Fig. 2)19. Anti-apoptotic members of the Bcl family, including Bcl-2 and Bcl-xL blunt intrinsic death signaling by blocking the recruitment of pro-apoptotic members to the mitochondria26. The cumulative data support the notion that high glucose might modulate the balance of the pro-apoptotic caspase family and anti-apoptotic Bcl proteins toward apoptosis, thus leading to β-cell death23,27.
One of immunocytochemical markers for apoptosis is cleaved caspase-3 (ACC): The caspase-3 protein is a member of the cysteine-aspartic acid protease (caspase) family and plays central roles in the execution-phase of cell apoptosis28. Caspases exist as zymogens in the soluble cytoplasm, endoplasmic reticulum, mitochondrial membrane space, and nuclear matrix22. Caspases are inactive proenzymes that undergo proteolytic processing at conserved aspartic residues to produce two subunits, large mass of ~20 kDa and small mass of~10 kDa, that dimerize to form the active enzyme19. This activated enzyme cleaves and activates caspase-6 and -7, and the protein itself is processed and activated by caspases- 8, -9 and -1029. One of these cleaved caspases is present on activated caspase-3, a ubiquitously distributed caspase which is a main effector caspase of the apoptotic cascade within cells29-33. Caspase-3 is active over a broad pH range that is more basic than many of other executioner caspases, indicating that caspase-3 will be fully active under normal and apoptotic cell condition29. Caspase-3 is activated in the both extrinsic and intrinsic apoptosis pathway. In the extrinsic pathway caspase-3 plays a dominant role by activating caspase cascade of apoptotic pathway. In the intrinsic pathway, cytochrome c from the mitochondria works in combination with caspase-9, apoptosis activating factor (Apar-1) and ATP to process procaspase30,31.
Immunocytochemical study
The commercially available polyclonal anti-ACC detects endogenous levels of the large (17/19 kDa) cleaved caspase-3 resulting from cleavage adjacent to Asp 175 and does not recognize the full length or other cleaved caspases (Cell Signaling Technology Publication, Beverly, MA, USA, 2006)34 and its immunopositive staining is specifically in the islet cell nucleus alone35,36. Recently, an involvement of caspase-3 in both T1DM and T2DM was implicated: in T1D, Fas(CD 95)-Fas L (CD 178) may be critical for β-cell destruction, as apoptosis in β-cell clone expressing the human Fas β-cell line is mediated by elevated caspase-3 like activity in tissue culture37 and the frequency of β-cell apoptosis in T2DM pancreatic tissues from autopsy is increased using deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)38.
We performed ACC immunocytochemical staining in 8 cases of T1D pancreata compared with 8 controls to study the converging point of extrinsic and intrinsic pathway19. T1DM islets showed more ACC-positive cells at 16% of the total islet cells, with large and small islets positive at 14% and 17%, respectively at 3.3 and 3.6 and 2.4 times that of the corresponding control total, large and small islet values, respectively (Table 1)39. The T1DM islets were a mixture of small and large regenerating islets consisting of non-β-cells of α-, δ- and pancreatic polypeptide (PP)-cells with moderately increased ACC-positive cells (Fig. 3A-E, right column) (Table 1)39. Among generally small islets from T1DM individuals (Case 6), which consist of insulin cells (25%) and more α-cells (75%), δ-cells (10%) and there were 13% ACC-positive β-cells at three times more than control islets (Table 1, Fig. 4A-D)39. The presence of increased ACC-cells in these islets suggest that these islets were in the process of increased β-cell apoptosis in the remaining β-cells. One type of large islets consists exclusively of α-cells and PP-cells (50% each) with practically no remaining β-cells and less ACC-cells, suggesting that these large islets correspond to the burnt-out, end-stage apoptosis-completed stage in response to long-stand compensation of insulin secretion (Fig. 4.E-H) (Case 8)39. Another type of large islets consists of remaining weakly insulin-staining minor β-cells, major α-cells and relatively decreased islet amyloid polypeptide (IAPP)-positive β-cells (16%), which represent remaining β-cells with less insulin and lesser IAPP immunostaining, thus suggesting apoptotic β-cell containing less insulin and even lesser IAPP, (Case 7, Table 2) (Fig. 5 A-D)40. The extra-large islets from Case 7 in Table 2 revealed weakly insulin-positive granular β-cells containing abundant cytoplasm with lesser IAPP-positive cells (17%), major α-cells (39%) and increased δ-cells (28%) plus irregular sickle-shaped densely IAPP-positive β-cells (Fig. 5 E-H)40. The pancreas was obtained at autopsy from T1D subjects of ages 18 to 75, and the immunocytochemical presence of ACC suggests that ACC mediated apoptosis was in the ongoing process even at the subjects’ death with a delicate balance of β-cell apoptosis and regeneration. These data also support that β-cell apoptosis and regeneration process is progressive from the preclinical stage to the end-stage of T1DM and T2D40,41. The ACC positive β-islet cells were more in type 1 diabetic islets than in type 2 diabetic islets39-41.
Table 1. Cleaved caspase-3 immunostaining in islets (total 30 islets for each case) from 8 T1DM subjects compared to 8cntrol subjects.
Numbers in parenthesis are the numbers of islets examined. *p values calculated with the corresponding control values are: p < 0.001.
Table 2. IAPP immunostaining for pancreatic islets from type 1 diabetics
Ins, Insulin; Glu, Glucagon; S RIF, Somtostatin; IAPP, Islet amyloid polypeptide; Ex large, Extra large islet; NC, Not calculable.
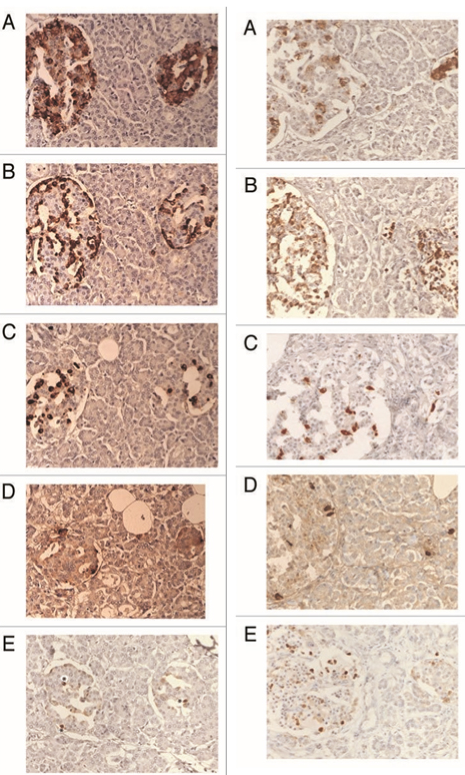
Figure 3. Control islets (left column) and T1D islets (right column).
Control islets consist of the major β-cells (up to 80%), α-cells (10%), δ-cells (<7%) and PP cells (<5%).Cleaved caspase-3 immunostaining was exclusively in the nucleus at about 5% of islet cells. In T1DM islets, residual β-cells, when they are present, are weaker immunostained than the control cells at 3-24% as the islets replaced by α-cells (up to 80%), disproportionally increased δ-cell and PP-cells with much increased nuclear positive cleaved caspase-3 staining in the corresponding β-cells. A: Insulin, B: Glucagon, C: Somatostatin, D: PP and E: Cleaved caspase-3 immunostained. From reference 34. Tomita, T, Islets 2010;2(1):24-29, with permission.
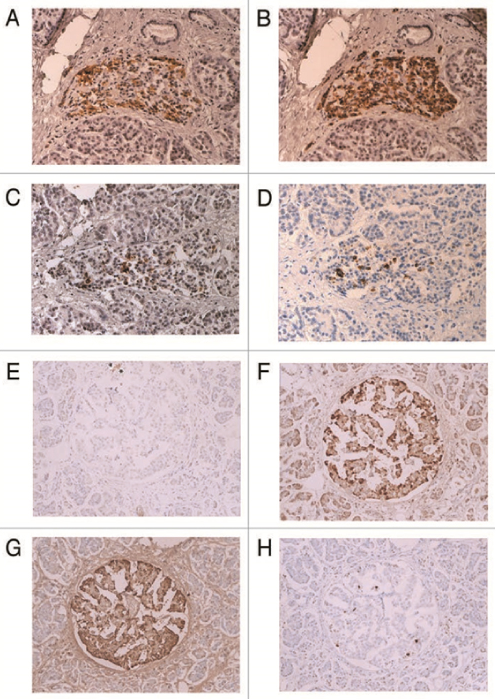
Figure 4. Insulin-poor small islet (A-E) and insulin-absent large islet (E-H) from T1D subjects in Table 1.
The islet from Case 6 (A-D) are generally small, consisting of insulin-cell poor (25%), glucagon-cell rich (75%) and somatostatin-cell rich (10%) with increased cleaved caspase-3 positive cells (13%). The large islet from Case 8 (E-H) -has practically no insulin-cells (<1%) and consists of major glucagon and pp-cells (a combined 100%) with a normal number of cleaved caspase-3 positive cells (4%). (*): Residual insulin-cells A and E: Insulin, B and F: Glucagon, C and G: PP, D and H: Cleaved caspase-3 immuno- stained. From reference 34, Tomita, T, Islets 2010; 2(1):24-29, with permission.
Butler and his associates studied type 1 diabetic islet using ACC and TUNEL immunofluore-scence: ACC immunostaining revealed 5.8% for type 1 diabetic islets compared to 2.7% for control islets at twice as much for type 1 diabetic islets of the control islets42. Our data on ACC-positive cells were 15.7 % for type 1 diabetic islets and 4.7% for control islets at three times more ACC-positive cells in type 1 diabetic islets than control islets39. Butler’s group reported TUNEL immunofluorescence at 0.2% for type 1 diabetic islets as compared to 0% for control islets43. Observing immunofluorescence photomicrographs, the positive staining was not clear and hazy for both ACC immunostaining and TUNEL including positive staining in the perinuclear cytoplasm42, not as clear as our ACC immunoperoxidase staining39,41. Another study using TUNEL reported 0.5% positive cells for normal β-cells43. Immunopositive ACC is present in the apoptotic cells and ongoing apoptotic cells39,41, which may explain a higher percentage of apoptotic cells revealed by ACC immunoperoxidase staining than TUNEL staining42,43. Thus, ACC immunoperoxidase staining recognizes protein levels more than TUNEL, the latter recognizes DNA levels42,43.
With 10 cases of T1D islets, immunocytochemical study was performed for insulin, glucagon, somatostatin (SRIF) and islet amyloid polypeptide (IAPP), the latter is a 37 amino acid polypeptide and is concomitantly co-secreted with insulin from β-cells into the blood stream in response to glucose -stimulated secretion40 : T1D islets were divided into insulin- absent islets from three patients younger than 40 years of age (Cases 1-3) and insulin-poor islets from seven patients older than 40 years of age (Cases 4-10) (Table 2)40. In three patients younger than 40 years of age, islets composed of β-cells with less than 16% of total islet cells or no-insulin positive cells in those two young subjects who died of diabetic ketoacidosis (Cases 2 and 3) where α-cells were major cells at 67% and the remaining somatostatin (SRIF) cells were 20---30% at twice the ratio of the control islets (Table 2)40. In Cases 6,7,9 and 10, α-cells were the major cells at 41—46% and β-cells were at 30—34% (Table 2)40. The four cases (Cases 6,7,9 and 10) revealed a mixture of extra-large, large and medium sized islets. The extra-large islets contained more than 100 islet cells, which were rare in in the control islets (Table 2)40. In large and medium sized islets, all β-, α- and δ-cells contained scanty cytoplasm and α-cells were more numerous than β-cells in majority of islets (Fig. 5 A, C and D)40. About 10% of type 1 diabetic islets were extra-large islets in Cases 6,7,9 and 10, in which majority were α-cells (Fig. 5G) and β-cells were weakly insulin-stained than the control β-cells (Fig. 5A and E)40. IAPP immunostai-ning was even weaker and less granular than insulin staining and some deeply IAPP positive staining was noted in sickle-shaped cytoplasm, often without nucleus, which appeared to be degenerating or dying β-cells (Fig. 5F)40. SRIF cells were relatively increased with abundant cytoplasm (Fig. 5H)40. Although β-cells from T1D subjects were much less in number and less immunostained for insulin than islets from T2D subjects, these residual β-cells could be activated or rejuvenated to produce and supply at least a part of endogenous insulin for better glucose homeostasis.
Lam et al studied 47 cases of T1D pancreata compared to 59 cases of nondiabetic controls for β-cell mass, β-cell turnover using Ki-67 and insulin/glucagon co-expression by immunocyto-chmical staining and TUNEL: residual insulin-producing β-cells were detected in some cases but not in all cases and several T1DM pancreata had substantial numbers of β-cells. β-cell proliferation was prominent early in life but it dramatically declined after infancy and β-cell proliferation by Ki-67 and β-cell proliferation was equivalent and β-cell death by apoptosis by TUNEL was rare in both T1D and control islets44. This study also supports the remaining insulin-positive β-cells as seen in our cases and rare detection of apoptotic β-cells using TUNEL44. Bressenot et al studied apoptosis in MDA-MB231 monolayer cells and HT29 human colon adenocarcinoma cell line using antibody against active caspase-3, active caspase-7 and cleaved poly-ADP-ribose polymerase 1 (c-PARP) immunofluorescent method: apoptosis-induced cells showed a significant higher number of active caspase-3-labaled cells, and active caspase-3 and c-PARP immunofluorescent perfectly colocalized in monolayer cells. A restricted expression of c-PARP was obvious in the greater part of active caspase-3 expressing cells45. There was absent caspase-7 immunofluorescence in some active caspase-3 positive cells and the relevance of each antibody for apoptosis needs to be further scrutinized45.
Molecular aspect on β-cell apoptosis
The Bcl-2 family governs mitochondrial outer membrane permeabilization and can be either anti-apoptotic (Bcl-2, Bcl-xL, Bcl-w) or pro-apoptotic (Bad, Bid, Bik, Bak) and there is a total of 25 genes known in the Bcl-2 family46,47. Bcl-2 family exerts anti- and pro-apoptotic effect by activation or inactivation of an inner mitochondrial permeability transition pore, which is involved in the regulation of matrix Ca2+, pH and voltage. Bcl-2 family proteins can induce by pro-apoptotic members or inhibit by anti-apoptotic members through the release of cytochrome c into the cytosol, which activates caspase-9 and -3, leading to apoptosis48. The members of the Bcl-2 family share one or more of the four characteristic domains (BH1, BH2, BH3 and BH4) and the anti-apoptotic Bcl-2 and Bcl-xL conserve all four BH domains49. All pro-apoptotic proteins contain a BH3 domain necessary for dimerization with other proteins of Bcl-2 family (Bax and Bak). The BH3-only of the Bcl-2 family of proteins only contains a single BH3-domain and plays a key role in promoting apoptosis. The BH3 -only family members are Bim, Bid and Bad49.
In the thymus, the elimination of potentially autoreactive T lymphocytes occurs by positive and negative selection, the latter involving activation-induced cell death (AICD) ensures an efficient elimination of T lymphocytes by apoptosis and this is a major mechanism in the regulation of peripheral tolerance and subsequently can determine the development of autoimmunity50. To evaluate the role of caspase-3 in T1D AICD, caspase-3 expression was analyzed in peripheral blood lymphocytes from 37 new onset T1D subjects and from 36 normal control subjects in the resting conditions and after anti-Fas triggered apoptosis. Caspase-3 mRNA expression was reduced in the resting lymphocytes in 18/37 T1DM subjects and 1/36 controls50. The T1D subjects studied from both Fas mediated AICD and caspase-3 mRNA expression revealed that a reduced caspase-3 mRNA expression in the resting lymphocytes in all T1DM subjects showing resistance to Fas mediated apoptosis50. Caspase-3 protein analysis confirmed mRNA data and showed an impaired expression of caspase-3 active form in T1DM subjects compared to the controls50. The defective expression and function of caspase-3 in peripheral lymphocytes in T1DM subjects may contribute to the development of AICD resistance in T1DM50.
With the right atrial appendages from diabetic and nondiabetic subjects retrieved during elective coronary bypass surgery, sliced appendages (300--500μm) were subjected to 90 min stimulated ischemia and 120 min reoxygenation51. The tissues were sectioned at 10 μm and were processed to TUNEL for apoptotic analysis. The apoptotic percentages were 12.5 ± 2.9 % and 15.0 ± 2.8 % for T1D and T2D groups, respectively, relative to 3.7 ± 0.8 % (p<0.05) in the nondiabetic groups51. The level of caspase activity in the cellular lysates was detected by fluorescence signal and the amount of acapase-3-like activity was measured using the effector caspase inhibitor zVAD-fmk. The activities of initiator caspases in fresh nondiabetic tissues were similar in T1D, T2D and nondiabetic groups, but the activity was significantly increased in T1DM and T2D groups relative to nondiabetic group after ischemia and re-oxygenation51. The caspase-3 inhibitor completely abolished apoptosis in the nondiabetic group but in T1DM and T2DM groups caspase-3 inhibition reduced apoptosis by 50%51. Thus, appendage tissues from T1DM and T2DM subjects revealed increased apoptosis after ischemia and re-oxygenation and caspase activities significantly increased in T1DM and T2DM tissues relative to nondiabetic group especially after ischemia and re-oxygenation and this 50% reduction of apoptosis by caspase-3 inhibitor was partially blocked51.
For a genetic aspect to study the role of caspase-3 in β-cells in vivo, the caspase-3 knockout (Caspase3-/- ) mice were tested: the knockout mice were protected from autoimmune diabetes by MLDS injection with 40 mg/kg of body weight, ip for 5 days , in which insulitis lymphocytic infiltrates in pancreatic islets was completely absent50. In Casp3+/- mice blood glucose was above 25 mmol/l compared to Casp3-/- mice whose blood glucose was below 10 mmol/l52. At 14 days post MLDS, pancreatic islets from Casp3+/- mice revealed less β-cells and predominant α-cells compared to Casp3-/- mouse islets consisting of major β-cells and less α-cells in the outer margin of the islet52. β-cell antigen-specific T-cell activation and proliferation were observed only in the pancreatic draining lymph node of RIP-GP/p14 Casp3+/- mice but not in Casp3-/-mice50. A genetic approach to transfer Acsp3+/- to Casp3-/ - may provide a future genetic engineering to stop the development of T1D52. c-Myc is a potent inducer of both cell proliferation and apoptosis and sensitizes cells to apoptotic triggers by augmenting the death receptor pathway and priming the mitochondria to release cytochrome c53. C-Myc has a predominantly pro-apoptotic role in pancreatic β-cells53. Woo and her associates further used the caspase-3 knockout mice containing inducible c-Myc transgene (c-Myc+Casp3-/-). C-Myc-activated Casp3-/- mice were protected from MLCD-diabetes, revealing that caspase-3 deletion confers protection from c-Myc-induced apoptosis and MLCD-diabetes53.
Islet transplantation can provide insulin independence in T1D subjects but most islet recipients revert to insulin dependence because up to 70% of transplanted islets lose adequate insulin secretion within the first week of transplantation53,54. Most islets do not function due to the host immune rejection, nonspecific inflammatory response and poor revascularization50,51.
Transfection of rat insulinoma cells (INS-1E cells) with chemically synthetic small RNA (siRNA) reduced caspase-3 transcripts by 50%55. Adv (replication deficient adenoviral) vector encoding shRNA transduced islets showed relatively prolonged levels of gene silencing and protected islets from cytokine-induced apoptosis56. Return to normoglycemia was achieved by transplanting Adv-caspase-3-shRNA transfected human islets under the kidney capsule in the MLDS mice53. INS-1E cells were further transfected with X-linked inhibitor of apoptosis (Adv-hXIP) which protects Ins-1E cells from inflammatory cytokines through decreasing the activities of caspase 3/7, 8 and 9, which resulted in reduced apoptotic β-cell65. Prolonged normoglycemia was achieved by transplanting Adv-XIP transfected human islets under the kidney capsule of the MLDS mice57. XIAP (X-linked inhibitor of apoptotic protein) of a potent endogenous inhibitor of downstream effector caspases 3,7 and 9 prevents apoptosis induced by both extrinsic and intrinsic signals57,58.
Emamaulee and her associates transplanted 500 and 250 islets under the kidney capsule in the MLDS mice and control mice59. Those treated with zVAD-fmk (10 mg/kg body weight) MLDS mice from day 0 to 5 posttransplant showed improved blood glucose levels compared with controls59. Ninety percent of zVAD-fmk -treated MLDS mice became euglycemic with 250 islets versus 27% of the control mice59. Short-term zVAD-fmk treatment significantly reduced posttransplant apoptosis in islet graft and thus pan-caspase inhibitor therapy is beneficial in promoting early marginal mass islet engrafting posttransplant59.
Glucose toxicity
To study glucose toxicity as a deleterious effect of chronic hyperglycemia on β-cells in vitro, Federici et al27 cultured 400 isolated human islets per batch for 5 days in a low glucose (5.5 mmol/l) and a high glucose medium (16.6 mmol;/l) for studying possible high glucose effects on Bcl-2 family gene expression by reverse transcription polymerase chain reaction27.
1) Bcl-2 was highly expressed in both low and high glucose media and expression did not change between a high and a low glucose condition (Fig. 5A)27.
2) however, Bcl-xL was reduced by 45% in the high glucose cultured islets compared with that of a low glucose condition, supporting that a reduction in Bcl-xL gene expression was due to high glucose exposure (Fig. 5B)27.
3) Bad, Bid and Bik were expressed in low glucose medium at low levels. Bad gene expression was greatly increased with a high glucose medium and Bad RNA level increased 80% as well compared with that of low level glucose (Fig. 5B and C)27. Bid gene expression was markedly increased with high glucose medium and so were Bik gene (Fig. 5D and E)27. Thus, anti-apoptotic Bcl-2 was unaffected by high glucose but pro-apoptotic genes, Bad, Bid and Bik markedly increased in high glucose cultured islets (Fig. 5)27. These data support that chronic high glucose incubation in vitro modulates the balance of pro-apoptotic and anti-apoptotic Bcl proteins toward apoptosis, thus leading to eventual β-cell death27.
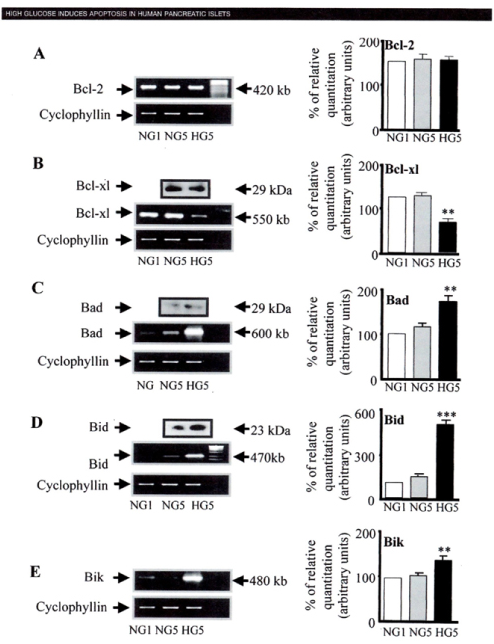
Figure 5. Large islet from 75 year-old female, Case 9 (A to D) and extra-large islet from 61 year - old female, Case 7 (E to H) in Table 2
In large islets, dark stained α-cells with small compact cytoplasm are major islet cells with relatively increased δ-cells whereas residual β-cells are weakly stained in the scanty cytoplasm compared with plump darkly stained cytoplasm of the control β-cells and IAPP immunostaining was even much weaker than insulin staining. In extra-large islet, there are a few scattered β- cells with weak, granular staining whereas there are abundant α-cells and increased δ-cell with a few corresponding β-cells, some of which showed dark sickle-shaped staining for IAPP (E to H). A and E: Insulin, B and F: IAPP, C and G: Glucagon, D and H: SRIF immunostained. From reference 39, Tomita, T, Islets 2011, 3-4(1):1-9, with permission.
Using MIN6N8 cells, which are SV40 transformed insulinoma cells derived from NDO mice, Kim et al extensively studied the effects of chronic hyperglycemia in the tissue culture: when MIN6N8 cells were cultured in different concentrations of glucose for varying time periods: a high glucose (33.3 mmol/l) induced marked genomic DNA fragmentation in a time- and dose-dependent manner and caused a significant increase of TUNEL-positive cells compared to a low glucose (5.5 mmol/l) cultured cells, concomitant cleavage of poly (ADP-ribose) polymerase (RARP) similar to caspase-3 cleavage (Fig. 6A)60. Pretreatment with a specific caspase-3 inhibitor (z-DEVD-CHO) completely reduced a high glucose-induced RARP cleavage (Fig. 6C)60 and apoptosis. Culturing with a high glucose significantly increased Fas and P53 expression whereas decreased Bcl-2 and Bcl-xL expression (Fig. 6B)60. A significant mitochondrial release of cytochrome c into cytosol was observed after 2 days (Fig. 6C)60. Translocation of Bax was confirmed by immunostaining: Bax interacted with VDAC (Fig. 6E)60 and Bax oligomerization substantially increased 2 days in a high glucose medium (Fig. 6F)60. Thus, chronic exposure to a high glucose increased through Bax oligomerization, cytochrome c release and caspase-3 activation, leading to β-cell apoptosis60. Glucose toxicity on β-cell apoptosis was studied with chronic exposure to a high glucose, which markedly reduced glucokinase (GCK, hexokinase IV) expression in a time- and dose-dependent manner61. Immunocytochemistry on tissue culture cells showed that glucose reduces GCK expression dose dependently decreasing by 40% in a high glucose medium. Chronic exposure to a high glucose for 4 days abolished to stimulate insulin content and inhibited ATP production62.
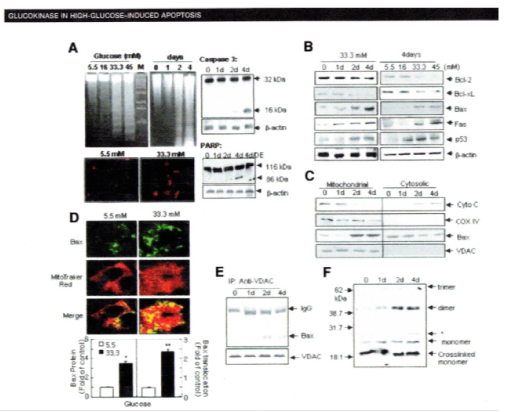
Figure 6. Bcl family gene regulation in human islets cultured in high versus normal glucose (A to E).
Expression of Bcxl-2, Bcl-xl, Bad, Bid and Bik mRNA was detected reverse transcription polymerase chain reaction (RT-PCR) and quantified by Fluorimager analysis of ethidium bromide signal. In each experiment, band densities were normalized against cyclophyllin, and the result are expressed mRNA level to NGI control islets (NGI=100%). A: Bcl-2, B: Bcl-xl, C: Bad, D: Bid, E: Bik From reference 22, Federici, M, Hrivbal, M, Perego, L et al. Diabetes 2001; 50(6):1290-301,with permission.
Main molecular studies of apoptotic Bcl proteins had been performed in artificially forced overexpression experiments in vitro62-64. Caspases are activated in a hierarchy order, in which initiator caspases (caspase-8 and -10) function to cleave effector caspases (caspase-3 and -7), the latter in turn degrade intercellular protein substrates and lead to the classical morphological changes of apoptosis (Fig. 2)19. Extracellular events present during the inflammatory response through the release of cytokines, including INF-σ, Il-1β and interferon-ϒ by infiltrating leukocytes or direct cytotoxic T-cell engagement, can initiate apoptosis18. These intrinsic cues function via surface molecule in the death receptor pathway, where specific ligand-receptor binding such as TNFα-TNF receptor binding, Fas -Fas L binding leads to receptor clustering, adaptor molecule recruitment and formation of death-inducing in signaling complex (DISC) (Fig. 2)19. Caspase-8 associates with DISC complex, where it is activated, released and leads to effector activation for caspase-346. Intracellular cues such as DNA damage, hypoxia, nutrient deprivation or reactive oxygen species (ROS) function via the mitochondrial pathway, which is tightly modulated by the Bcl-2 proteins (Fig. 6)31,60. In healthy cells, pro-apoptotic Bcl-2 proteins (Bim, Bid, Bad, Bax and Bak) are present in the mature form, while anti-apoptotic Bcl-2 proteins (Bcl-2 and Bcl-xL) are constitutively active and reside in the outer membrane of mitochondria (Fig. 6)31,60. Following an intrinsic cue, proapoptotic Bcl proteins become activated and translocate to the mitochondria, where they bind to inactivate anti-apoptotic Bcl-2 proteins or form pores in the mitochondrial membrane, which facilitates the release of cytochrome c into the cytosol (Fig. 2)19. When cytochrome c accumulates in the cytosol, it complexes with procaspase-9 and Apaf-1 to form the “apoptosome”, which in turn activates caspase-3 (Fig. 2)19,61-63. Both intrinsic and extrinsic signaling cascades converge at the point of caspase-3 activation, which is often considered as the “point of no return” in apoptosis18. Apoptosis can only occur when the concentration of pro-apoptotic Bcl proteins exceeds that of anti-apoptotic proteins at the mitochondrial membrane of the intrinsic pathway27. Using transgenic mice, Daniel el al found: glucose-induced changes in the mitochondrial membrane potential were significantly reduced in Bad-/- β-cells and the average (Ca 2+) I response to 11 mM glucose was significantly lower in Bad-/-β-cells65,66. An intact BH3 domain is required for glucose-stimulated insulin secretion by its binding to Bcl-2 and Bcl-xL. Treatment with BAD SAHBA (stabilized α-helix of Bcl-2 domain) restored the secretion defect in Bad -/- islets, underscoring the sequence specificity of the BAD SAHB effect66. α-helical destabilization of the Bcl-2-BH4-domain abolishes its ability to inhibit the IP3 receptor67. They further identified GCK as a novel and direct physiological target of the BAD BH3 domain in β-cells and that phosphorylation within the BH3 domain drives the metabolic functionality of BAS and severs as a physiological switch of its apoptotic and metabolic effects66.
The early event of β-cell dysfunction includes endoplasmic reticulum stress and oxidative stress68 leading to β-cell exhaustion and eventual apoptosis. Pancreatic islets represent only 2% of the pancreas but produce the most important hormone for survival, insulin for glucose homeostasis, in which a human adult needs about 40 Unit of insulin (about 13 mg) per day. Endoplasmic reticulum (ER) is an organelle responsible for protein folding and assembly and transport of synthesized protein in cisternae67. The ER is well developed in β-cells for synthesizing large amounts of insulin69. Rough ER (RER) is in charge of protein synthesis and the newly synthesized proteins are folded into three-dimensional structure in RER and is sent to Golgi complex or membrane via small vesicles69. ER stress, the cellular responses to the disturbance of normal function of ER, is the most common cause for protein miss-folding. In T1DM, increased insulin synthesis in residual β-cells exceeds the folding capacity of ER, resulting in the accumulation of unfolded protein. The unfolded proteins including insulin, are prone to aggregate with each other in a crowded environment and directed to degradative pathway, which further exacerbate existing T1D69,70.
Thus, In the all intricate cascade of islet apoptosis, the therapeutic application of BAD SAHBs and other BAD SAHB that activates glucose-stimulated insulin secretion, but does not affect the survival function of Bcl-xL, may serve as a prototype therapeutic application in diabetes and islet transplantation69.
Conclusion
As mentioned so far, there are a milliard of cascade of biological processes for β-cell destruction by an autoimmune reaction in T1D while β-cells are replaced by non-β-cells at a delicate balance of islet cell apoptosis and regeneration in long time frame. Since other environmental factors are involved for the development of T1D in addition to genetic factors, there are many ways to theoretically and eventually prevent or retard autoimmune reactions in the complicated processes of T1D development. Restore insulin secretion on the remaining non-functioning β-cells in T1D may lead to rejuvenate and activate production and secretion of more endogenous insulin. It is also evident that an adequate glucose homeostasis in T1D is a prudent way to protect the remaining β-cells in T1D subjects against hyperglycemia-induced β-cell apoptosis. There is a future hope for therapeutic application to slow or even prevent β-cells apoptosis pathways in T1D at the molecular level by deleting Casp3+/- to Casp3-/- and silencing Casp3+/+ gene or using pan-caspase inhibitor treatment to prevent and slow progressive apoptosis in pancreatic β-cells.
References
- Daneman, D. Type 1 diabetes: Lancet. 2006; 367 (9513): 847-858.
- Yoon JW, Jun HS. Autoimmune destruction of pancreatic beta-cells. Am J Ther. 2005; 12(6):580-591.
- Pirot P, Cardozo AK, Eizirk DL. Mediators and mechanisms of pancreatic beta-cell death in type 1 diabetes. Arq Bras Endocrine Meatv. 2008; 52(2):156-165.
- Jahromi, MM, Eisenbarth, GS. Cellular and molecular pathogenesis of type 1A diabetes. Cell Mol Life Sci. 2007; 64 (2): 865-872.
- Cnop, M, Welsh, N, Jonas, JC, et al. Mechanisms o pancreatic beta-cell death intype 1 and type 2 diabetes: many differences, few similarities’. Diabetes. 2005; 54(Suppl 2): s97-s107.
- Kim MS, Polychronakos, C. Immunogenetics of type 12 diabetes. Horm Res. 2005; 64(4):180-188.
- Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007; 39(6): 857-864.
- Turner D. The human leucocyte antigen (HLA) system. Vox Sang. 2004; 87(suppl1):87:87-90.
- Pihoker C, Gilliam LK, Hampe CS, et al. Autoantibodies in diabetes. Diabetes. 2005; 54(Suppl 2): s52-s611.
- Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transport ZnT8 (SLc30A8) is a major autoantigen in human type 1 diabetes. Pro Natl Acad Sci USA. 2007: 104(43): 17040-17045.
- Filippi CM, von Herrah MG. Viral trigger for type 1 diabetes. Diabetes. 2008; 57(11): 2863-2871.
- In’t Veld P, Lievens D, De Grijse J, et al. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes. 2007; 56(9): 2400-2004.
- Knip M, Veijola R, Virtanen SM, et al. Environmental triggers and determinants of type 1 diabetes. Diabetes. 2005; 54 (Suppl2): s125-s136.
- Wilcox A, Richardson SJ, Bone AJ, et al. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2008; 155(2):173-181.
- Pugliese A. Autoreactive T cell in type 1 diabetes. J Clin Invest. 2017; 127(8): 2881-2891.
- Itoh N, Hanafusa A, Miyazaki A, et al. Mononuclear cell infiltration and its relation to the expression of major complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993; 92(5): 2313-2322.
- Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity. 2010; 32(4): 468-478.
- Prause M, Christensen DP, Billestrup N, et al. JNK1 protects against glucolipotoxicity-mediated beta-cell apoptosis. PLOS One 2014, 9(1): e87067.
- Lee SC, Pervaiz S. Apoptosis in pathophysiology of diabetes mellitus. Int J Biochem. 2007; 39 (3): 497-504.
- Nakano M, Matsumoto I, Sawada T, et al. Caspase-3 inhibitor prevents apoptosis of human islets immediately after isolation and improves islet graft function. Pancreas. 2004; 29 (2): 104-109.
- Cheng G, Ahu L, Mahato RI. Caspase gene silencing for inhibiting apoptosis in insulin cells and human islets. Molec Pharmac. 2008; 5(6): 1093-1102.
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997; 22(8): 299-306.
- Chandra J, Zhivotovsky B, Zaitsev, S, et al. Role of apoptosis in pancreatic β-cell death in diabetes. Diabetes. 2001; 50 (Suppl 1): s44-s47.
- Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998. 10(5): 545-551.
- Gross A, McDonnell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Gene Dev. 1999; 13(15): 1899-1911.
- Green D. Apoptotic pathways. Ten minutes to death. Cell. 2005; 121(5): 671-674.
- Federici M, Hrivbal M, Perego L, et al: High glucose causes apoptosis in cultured human pancreatic islets of Langerhans. A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001; 50(6): 1290-1301.
- Alnemri ES, Livingston DJ, Nicholson DW, et al. Human ICE/CED-3 protease nomenclature. Cell. 1996; 87(2): 171.
- Stennicke HR, Salvesen GS. Biochemical characteristics of caspses-3, -6, -7 and -8. J Bilol Chem. 1997; 272(41): 25719-25723.
- Katunuma N, Matsui A, Le QT, et al. Novel procaspase-3 activating cascade mediated by lysoapoptases and its biological significances in apoptosis. Adv Enzyme Regul. 2001; 41(5): 237-250.
- Li P, Nijhawn D, Wang X. Mitochondrial activation of apoptosis. Cell. 2004; 116(Suppl 21): s57-s59.
- Hirata H, Takahashi A, Kobayashi S, et al. Caspases are activated in a branched protease cascade and control distinct down-stream processes in Fas-induced apoptosis. J Exp Med. 1998; 187(4): 587-600.
- Tewari M, Quan LT, O’Rourke K, et al. Yama/ cp32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase. Cell. 1995; 81 (5): 801-809.
- Cleaved caspase-3 (Asp 175) antibody (2006) vol 9661, Cell Signaling Technology, Beverly, MA, Cat # 9661.
- Tomita T. Caspase-3 immunocytochemical staining for pancreatic islets and pancreatic endocrine tumors. Hum Pathol. 2009; 40(7): 1050-1056.
- Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: Immunohistochemistry using antibodies to cleaved-caspase3. J Histochem Cytochem. 2002; 50(4): 449-454.
- Martin SJ, Green DR. Protease activation during apoptosis: death by thousand cuts. Cell. 1995; 82(3): 349-452.
- Butler AE, Janson J, Bonner-Weir S, et al. β cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003; 52 (1): 102-110.
- Tomita T. Immunocytochemical localization of cleaved casspase-3 in pancreatic islets from type 1 diabetic subjects. Islets. 2010; 2 (1): 24-29.
- Tomita T. Immunocytochemical localization of caspase-3 in pancreatic islets from type 2 diabetic subjects. Pathology. 2010; 42(5): 432-437.
- Tomita T. Islet amyloid polypeptide in pancreatic islets from type 1 diabetic subjects. Islets. 2011; 3-4 (1): 1-9.
- Meir JJ, Bhushan A, Butler AE, et al. Sustained beta cell apoptosis with long-standing type 1 diabetes: indirect evidence for islet regeneration. Diabetologia. 2005; 48(11): 2221-2228.
- Butler AE, Galasso R, Meier JJ, et al. Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia. 2007; 50(11): 2323-2331.
- Lam CJ, Jacobson DR, Rankin M, et al. β-cells persist in T1D pancreata without evidence of ongoing turnover or neogenesis. J Clin Endocrinol Metab. 2017; 102: 2647-2659.
- Bressenot A, Marchal S, Bezdetnaya L, et at l. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7 or cleaved PARP in monolayer cells and spheroid and subcutaneous Xenografts of human carcinoma. J Histochem Cytochem. 2009; 57: 289-3000.
- Rhodes CJ. Type 2 diabetes-a matter of α-cell life and death. Science. 2005; 307(5708): 380-384.
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998; 281(5381): 1322-1326.
- Zamzami N, Brenner C, Marzo I, et al. Subcellular and submitochondrial mode of action of Bcl-2 like oncoproteins. Oncogene. 1998; 16(8): 2265-2282.
- Petros AM, Oleijniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochem Biophys Acta. 2004; 1644(2-3): 83-94.
- Vendrama F, Santangelo C, Misasi R, et al. Defective lymphocyte caspase-3 expression in type 1 diabetes mellitus. Eur J Endocrinol. 2005; 152(1): 119-125.
- Chowdhry MF, Vohra HA, Galinanes M. Diabetes increases apoptosis and necrosis in both ischemic and poly-adenosine diphosphate-ribose polymerase. J Thor Cardiovas Surg. 2007; 134(1): 124-131.
- Liadis N, Murakami K, Eweida M, et al: Caspase-3-dependent β-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. 2005; 25(9): 3620-3629.
- Radziszewska A, Schroer SA, Choi D, et al. Absence of caspase-3 protects pancreatic β-cell from c-Myc-induced apoptosis without leading to tumor formation. J Biol Chem. 2009; 284(16): 10047-10056.
- Choi D, Woo M. Execution of apoptosis in pancreatic β-cells: Not just for cell death. Am J Physiol Endocrinol. 2010; 298: E735-E741.
- Brissova M, Shostak A, Shiota M, et al: Pancreatic islet production of vascular endothelial growth factor-A is essential for islet vascularization, revascularization, and function. Diabetes. 2006; 55(11): 2974-2985.
- Emamaullee JA, Stanton L, Schur C, et al. Caspase inhibitor therapy enhances marginal mass islet graft survival and preserves long-term function in islet transplantation. Diabetes. 2007; 56 (5): 1289-1298.
- Cheng G, Zhu L, Mahato RI. Caspase-3 silencing for inhibiting apoptosis in insulinoma cells and human islets. Mol Pharmaceut. 2008; 5(6): 1093-1102.
- Wu H, Panakanti R, Li F, et al. XIAP gene expression protects β-cells and human islets from apoptotic cell death. Mol Pharm. 2010, 7(5): 1655-1666.
- Emamaullee JA, Rajotte RV, Liston P, et al. XIAP overexpression in human islets prevents early postransplant apoptosis and reduces the islet mass needed to treat diabetes. Diabetes. 2005; 54 (9): 2541-2548.
- Kim WH, Lee JW, Suh YH, et al. Exposure to chronic high glucose induces β-cell apoptosis through decreased interaction of glucokinase with mitochondria. Diabetes. 2005; 54(9): 2602-2611.
- Chan SI, Yu VC. Proteins of the Bcl-2 family in apoptosis signaling: from mechanic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol. 2004; 31(3): 119-128.
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000; 407 (6805): 770-776.
- Ou D, Wang X, Metzger DL, et al: Synergetic inhibition of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human pancreatic beta cells by Bcl-2 and X-linked inhibitor of apoptosis. Hum Immunol. 2005; 66 (3): 274-284.
- Saldeen J. Cytokines induce both necrosis and apoptosis via a common Bcl-2-inhibitable pathway in rat insulin-producing cells. Endocrinology. 2000; 141(6): 2003-2010.
- Daniel NN, Gramm CF, Scorrano L, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003; 424(6951): 952-956.
- Daniel NN, Walensky LD, Zhang CY, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nature Med. 2008; 14(2): 144-153.
- Monaco G, Decrock E, Nuyts K, et al: Alpha-helical destabilization of the Bcl-2-BH domain peptide abolishes its ability to inherit the IP3 receptor. PLOS One. 2013; 81(1): e73386.
- Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes. Inter Med. 2003; 42(1): 7-14.
- Zhong J. Endoplasmic reticulum (ER) stress in the pathogenesis of type 1 diabetes. In: Medicine: Endocrinology and Metabolism, Type 1 diabetes. Chapter 8, Ed. AP Fisher, A. Li, Intech, 2013; p 195-221.
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes. Endocr Rev. 2008, 29(1): 42-61.