
Fetal and neonatal alloimmune thrombocytopenia: Novel mechanisms of miscarriage learned from placental pathology in animal models
Issaka Yougbaré1-3, Darko Zdravic1-4, Heyu Ni1-6*
1*Toronto Platelet Immunobiology Group
2Department of Laboratory Medicine, Keenan Research Centre for Biomedical Science, St. Michael's Hospital, Toronto, ON Canada
3Canadian Blood Services Centre for Innovation, Toronto, ON K1G 4J5, Canada
4Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON M5S 1A8, Canada
5Department of Physiology, University of Toronto, Toronto, ON M5S 1A8, Canada
6Department of Medicine, University of Toronto, Toronto, ON M5S 1A8
Abstract
Fetal/neonatal alloimmune thrombocytopenia (FNAIT) is a life-threatening disease. Maternal alloimmune responses against fetal platelet antigens in FNAIT may lead to clinical complications including bleeding disorders, intrauterine growth restriction (IUGR) and in severe cases fetal death (miscarriage). It has been long suspected that thrombocytopenia may be the reason for bleeding disorders in FNAIT, recent studies from us and other groups, however, suggested that the anti-angiogenic effects of anti-platelet antibodies may play a key role in bleeding, particularly in intracranial hemorrhages. Our earlier studies using murine models also suggested that some anti-platelet antibodies can activate platelets and initiate thrombotic events in the placenta, which may contribute to miscarriage. Most recently, we found that maternal anti-β3 integrin antibodies can target fetal allogenic trophoblasts, form immune complexes, and generate binding sites for natural killer (NK) cell Fcγ receptors. Uterine NK cell activation through NKp46 and perforin release caused trophoblast apoptosis, impaired spiral artery remodeling, and ultimately lead to IUGR and/or fetal death. We found that NK cell-mediated placental pathologies are preventable by anti-NK antibody treatments, which may have translational importance. This mini-review mainly discussed the latest discoveries regarding activated uterine NK cells-mediated miscarriage. Future research on placental inflammation and remodeling should open new avenues for interventions in FNAIT-mediated pregnancy failures.
Background
Fetal/neonatal alloimmune thrombocytopenia (FNAIT) is a severe gestational disease in which the maternal immune system targets fetal platelet antigens1-5. Clinical complications include bleeding disorders ranging from mild cutaneous petechial to severe intracranial hemorrhages (ICH), intrauterine growth restriction (IUGR), and fetal or neonatal death2,5-9. In contrast to its analog of red blood cell alloimmunization, hemolytic disease of the newborn, FNAIT cases can occur during the first pregnancy10,11. The rate of recurrence among subsequent human platelet antigen (HPA)-positive siblings is almost 100%, with an increased disease severity in most of cases. In neonatal intensive care units, FNAIT is the most frequent cause of severe thrombocytopenia in neonates2. FNAIT has been reported to occur in 0.5-1.5 per 1000 live-born neonates. However, this may underestimate the incidence of FNAIT since in utero fetal death has not been well studied. The incidence and mechanisms of miscarriages in FNAIT are largely unknown.
Platelet glycoproteins (GP) are remarkably polymorphic receptors for key hemostatic factors, such as fibrinogen, von Willebrand factor, and collagen12-14. GPIIbIIIa (integrin αIIbβ3) and GPIbα are also targeted by the maternal immune system during pregnancy as well as in autoimmune diseases2,15-19. Polymorphic incompatibilities in human platelet antigens (HPAs) between the mother and fetus can trigger maternal alloimmune responses, which may be enhanced if infection co-exists19. At least 37 HPAs have been reported to cause FNAIT, and approximately half are located on the extracellular domain of GPIIIa (integrin β3 subunit)20. Recent studies estimated that in European descent, 85% of FNAIT cases are due to alloantibodies targeting HPA-1a which is due to a gene polymorphism in residue 33 (L33P) in β3 subunit20. HPA-5a (residue 505 Lys (K505E) on the GPIa (α2 subunit of integrin α2β1, the collagen receptor) alloimmunization is the second leading cause of FNAIT in the Caucasian population and those against other HPAs (5 %)10,21. In Asians, anti-HPA 4 (residue 143 (Q143R)) and anti-CD36 alloimmunizations are relatively common22.
Transplacental passage of maternal anti-platelet IgG antibodies to the fetus via neonatal Fc receptor (FcRn) transport causes fetal platelet opsonization and destruction, leading to thrombocytopenia23,24. Intracranial hemorrhage may occur as early as 14-16 weeks of gestation, which can be diagnosed by routine ultrasound follow-up of the pregnancy. It has been reported that up to 10% of live-born neonates with FNAIT had ICH in utero before the 30th week25. Postnatal ICH is also frequent (10-20%) in FNAIT neonates, and may lead to lifelong disabilities or death in up to 5% of cases9,25. It has been well recognized that maternal anti-β3 integrin alloantibodies are the major cause of the clinical manifestations of ICH.
Platelet alloimmunization and mechanism of intracranial hemorrhage
Platelets are small, versatile blood cells that play vital roles in hemostasis and thrombosis12-14,26,27. Therefore thrombocytopenia has been long suspected to be the major cause of ICH in FNAIT. We have recently demonstrated in our mouse model of FNAIT that impairment of angiogenesis can lead to ICH28. Anti-β3 integrin antibodies, by cross-reacting with αVβ3 on endothelial cells (ECs), induced apoptosis of ECs, particularly those tip cells. Tip cells are the leading cells of the sprouts of new blood vessels29. During angiogenesis, tip cells up-regulate αVβ3 integrin which is essential for cell migration. Disruption in αVβ3 integrin signaling has been reported to cause impaired vascularization and hemorrhage in several animal models29-31. This cross-reactivity between maternal antibodies and β3 integrins (i.e. αIIbβ3 and αVβ3) leads to decreased vessel density in affected brains and retinas.
Both murine anti-β3 antisera and human anti–HPA-1a IgG inhibit EC proliferation and in vitro angiogenesis by a mechanism involving Akt signaling. Other studies by Santoso and colleagues confirmed that anti-HPA-1a antibodies, which have a stronger binding affinity to EC-derived integrin αVβ3, may be predictive of ICH, and can be used as a guide to prophylactic treatment during pregnancy32,33. In a large international collaborative study of more than 600 cases of FNAIT, it was reported that non-invasive management using IVIG with or without additional steroids could lead to a favorable outcome, and prevent bleeding in the fetus or neonate4. Furthermore, experts in the field recommend weekly IVIG administration with or without corticosteroids, as a first-line antenatal strategy for the management of FNAIT34. Invasive methods such as fetal blood sampling, and in utero platelet transfusion have no significant benefit, therefore should be avoided32.
In those who survived ICH, neurological and vision impairment are other developmental concerns during childhood25. Lack of proper brain and retinal vascularization in neonates may explain the developmental origins of mental and vision abnormalities in FNAIT affected patients in their adulthood. This observation is supported by the fact that exposure to maternal anti-HPA-1a IgG has been reported to reduce birth weight in male children35.
Anti-platelet antibodies, impaired vascularization and in utero fetal growth restriction
In addition to being expressed on platelets and ECs, the most targeted antigen in FNAIT, β3 integrin is expressed on conceptus-derived trophoblast (placental) cells including extravillous trophoblasts (EVT), and syncytiotrophoblasts (ST)36,37. Trophoblast αIIbβ3 and αVβ3 integrins are early contributors to blastocyst implantation and subsequent placental development including spiral artery remodeling36. Invasive trophoblast cells replace existing vascular ECs in the placental spiral arteries, which lowers maternal vascular resistance and increases uteroplacental blood flow38. Inadequate migration and invasion of trophoblasts have been reported in complications of pregnancy, including miscarriage, preeclampsia, and IUGR39. Therefore, in addition to causing ICH, it is possible that maternal anti-β3 antibody may target αVβ3 integrin on trophoblasts, and impair placental angiogenesis and development leading to placental dysfunction, IUGR, and even miscarriage, in FNAIT (Figure 1).
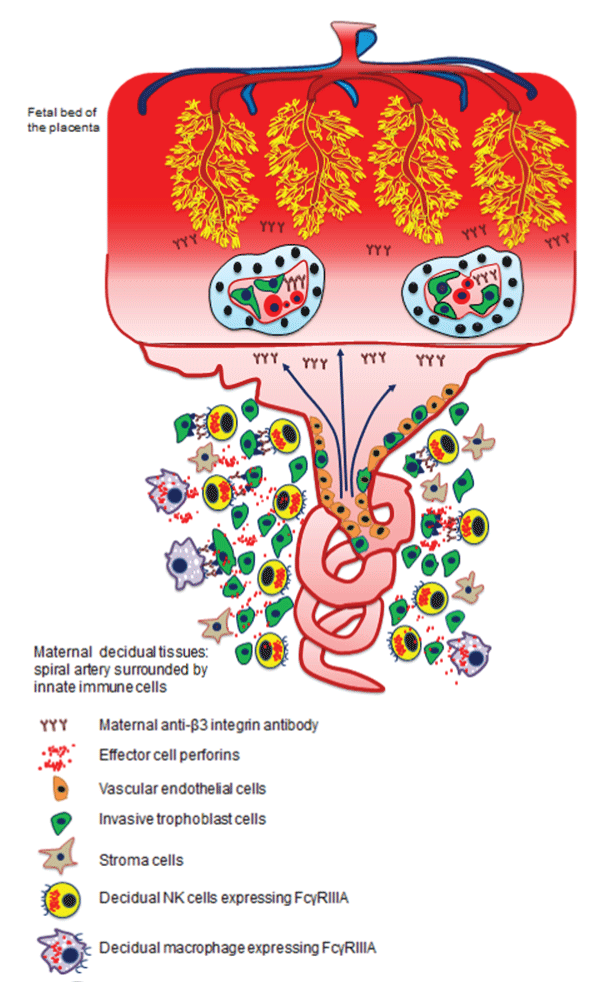
Figure 1:Mechanism of maternal anti- β3 integrin antibody and innate immune cells induced miscarriage in FNAIT. Maternal anti-β3 integrin form immune complexes on invasive trophoblasts where their Fc portion are exposed and generate binding sites for FcγRIIIA expressing cells. Uterine NK cell activation through NKp46 and perforin release induced trophoblast apoptosis and impaired spiral artery remodeling. Anti-β3 integrin antibodies cross placental blood barrier trough neonatal FcR transportation. These antibodies can bind to syncytiotrophoblast and cytotrophoblast expressing αVβ3 integrin thereby damaging blood barrier as well as affecting IgG molecules transport. Capillary development in the fetal bed is also impaired by maternal anti- β3 integrin antibodies.
Our recent findings published in Nature Communications1 shed light on the newly described mechanisms of growth restriction and fetal death due to placental abnormalities in FNAIT40. Intensive angiogenesis is required for proper sponge capillary development in the fetal bed of the placenta. In our murine model of FNAIT, impaired feto-placental blood flow and reduced heart rate were documented by ultrasound as signs of fetal distress before death. Maternal anti-β3 integrin antibodies, by cross-reacting with αVβ3 on ECs, caused reduced placental vascularization similar to other highly angiogenic organs during development such as brain and retina, as we previously reported28. Overall, EC and trophoblast proliferation was significantly reduced in affected placentas. It is understandable that these pathological features can limit nutrient transport from the maternal to the fetal compartment and in turn cause severe IUGR (Figure 1). Poor placental revascularization was accompanied by apoptosis of invasive trophoblasts, mediated by decidual natural killer cells at the feto-maternal interface.
Maternal immune response at the feto-maternal interface in FNAIT
A better understanding of placental anatomy and the maternal immune response at the feto-maternal interface may provide new insights into pregnant women’s tolerance of their semi-allogeneic fetus in FNAIT. Important factors contributing to the protection of the conceptus from the maternal immune system include: (i) the anatomical barrier that separates fetal and maternal circulation, (ii) reduced major histocompatibility antigen expression on trophoblast cells; and (iii) maintenance of an immunosuppressive environment within the uterus41 etc. For instance, the separation of the maternal/ fetal circulatory systems by the syncytiotrophoblast layer in the placental villi allows the fetus to grow as an isolated allograft completely protected from maternal immune cells. Syncytiotrophoblasts and invasive extravillous trophoblasts which come in close contact with maternal immune cells, escape allo-recognition because they lack classical human leucocyte antigen (HLA) class I and II molecules42.
The placenta is an immunologically privileged organ, and the mother does not ordinarily make pathogenic antibodies against the syncytiotrophoblast layer, even though it may express foreign, paternally derived antigens on its surface. However, this prevailing view in FNAIT can be challenged since maternal anti-platelet antibodies cross-react with αVβ3 integrin on trophoblasts. In addition, prostaglandin E2 and indolamine-2 produced by trophoblast cells, as well as T helper cell type 2 cytokines orchestrate the composition and regulatory function of maternal immune cells.
In early gestation, maternal immune cells in the decidua consist of uterine natural killer (uNK) cells (70%), macrophages (20%), T cells (9%), and few dendritic and B cells41,43. Among innate lymphoid cells (ILCs), NK cells are highly-enriched, transient lymphocytes that promote decidualization, including immune tolerance and vascular development44. As the main drivers of angiogenesis, the importance of NK cells in healthy, normal pregnancy has been demonstrated in pregnant mice devoid of NK cells. In addition to decidual NK cells, other innate lymphoid cells subsets have been recently reported including lymphoid tissue inducer (LTi)-like cells, natural cytotoxicity receptors (NCRs)+ ILC3, and interferon-(IFN)γ-producing ILC145. These newly described ILCs have been reported to play a role in innate defenses and vascularization as well as tissue remodeling during pregnancy. Uterine NK cells and trophoblasts are functional partners during pregnancy. Tightly regulated interactions between NK cells and trophoblasts dictate normal spiral artery remodeling as dNK cells prevent excessive invasion of trophoblasts, and trophoblasts inhibit the maternal immune response and fetal rejection46,47. In the maternal decidual tissues, infiltrated macrophages and NKs are the effector cells expressing in Fcγ Receptor IIIA and capable of inducing antibody-dependent cell-mediated cytotoxicity (ADCC). Although human decidual CD56+CD16- NK cells do not express FcγRIIIA during normal pregnancy, a phenotypic change may occur during inflammation. However, how inflammation and chemokines can change the expression of CD16 on dNK cells, and how chemotactic migration of peripheral CD16+ NK cell migration into to the decidua require further investigations. A recent study has shown that women with increased numbers of CD16+ uterine NK cells in the deciduas may be at greater risk of developing infertility disorders48. In addition it has been reported that peripheral blood NK cell recruitment to the uterus contributes to the accumulation of NK cells during early pregnancy48,49. Decidual macrophages may express FcγRIIIA, and are the second most abundant population of cells in the fetal-maternal interface. An increased number of decidual macrophages has been correlated with recurrent miscarriage. Efficient spiral artery remodeling is a coordinated activity between extravillous trophoblast cells, decidual uterine NK cells, and macrophages in a carefully, spatiotemporally regulated manner50. These findings support the hypothesis that cells expressing FcγRIIIA may be involved in ADCC in human FNAIT. It is highly possible that activated macrophages promote NK cell cytotoxicity during placental remodeling in FNAIT.
Immuno-inflammatory responses at fetomaternal interface in FNAIT
We recently reported that placental inflammation (decidual enlargement, cytokine profiles, and NK cell hypercellularity with aberrant activation and prolonged survival in decidua basalis) has a profound impact on fetal survival in FNAIT1. Previous clinical studies reported that IUGR and fetal demise occur as frequently as ICH in FNAIT51. A recent retrospective cohort study of 21 cases of human FNAIT reported signs of chronic inflammation at the maternal-fetal interface. Dubruc and colleagues documented that FNAIT was associated with a significantly higher frequency of chronic chorioamnionitis, basal chronic villitis, and chronic intervillositis. Furthermore, chronic villitis and chronic deciduitis were also more frequent in FNAIT cases. However, the subtypes of immune cells causing placental inflammation, and the mechanism by which trophoblast apoptosis occurs was not previously elucidated. We provide strong evidence that maternal alloantibody associated placental pathologies dictate not only the severity of bleeding disorders in fetuses and neonates but more importantly the severity of IUGR and likelihood fetal death. Deposition of maternal anti-β3 antibodies in the decidua initiates recruitment of post-mitotic NK cells from the peripheral circulation to the decidua. Indeed, the immune complexes trigger NK ADCC on invasive trophoblasts, trophoblast cell death, and subsequent pregnancy failure (Figure 1). Interestingly, we also found several therapeutic strategies can prevent NK cell-mediated placental pathologies including anti-NKp46, anti-Fcγ Receptor IIIA antibodies, and anti-asialo-GM-1 mediated NK cell depletion. This newly described mechanism and therapeutic approach targeting NK cells to prevent miscarriage may have great translational importance.
More research is needed to identify the phenotype of decidual immune cell composition involved in placental inflammation in FNAIT. Bleeding disorders and ICH have captured almost all of the attention for clinical management in FNAIT, yet little is known about placental abnormalities and miscarriages. Trophoblasts are unique cells in the placenta which are capable of cross-reacting with maternal antibodies and immune cells. It is likely possible that these are the first cells to be targeted and damaged by anti-HPA-1a antibodies. In this regard, it has been reported that anti-HPA-1a (mAb 26.4) antibodies affected trophoblast adhesion and migration, which are crucial for normal placental development52. Trophoblast apoptosis and an impaired placental blood barrier can affect IgG transport via the FcRn across the placenta.
The syncytiotrophoblast layer is an absorptive epithelium and has the important role of transport including IgG transplacental passage. It is unknown whether IgG or other immunoglobulin transport could be increased in FNAIT because of syncytiotrophoblast layer impairment. In this case, preventing placental lesions and inflammation could improve FNAIT induced IUGR, miscarriage and bleeding disorders.
Conclusion
FNAIT is a life-threatening disease. Devastating ICH may severely affect the brain and neurological development in neonates who survived FNAIT. The anti-angiogenic effects on retinal vessels observed in our model may also lead to vision impairment, although more clinical studies are necessary to confirm this hypothesis. Reciprocally, on the maternal side, placental insufficiency due to impaired spiral artery remodeling may lead to complications of pregnancy and recurrent miscarriages in FNAIT. Future research in FNAIT devoted to placental abnormalities, inflammation mediated by decidual immune cells, and the integrity of the placental blood barrier may lead to new avenues for clinical management and therapy development.
References
- Yougbare I, Tai WS, Zdravic D, et al. Activated NK cells cause placental dysfunction and miscarriages in fetal alloimmune thrombocytopenia. Nat Commun. 2017; 8: 224.
- Zdravic D. Fetal and neonatal alloimmune thrombocytopenia. Semin Fetal Neonatal Med.(2016; 21: 19-27.
- Vadasz B, Chen P, Yougbaré I, et al. Platelets and platelet alloantigens: Lessons from human patients and animal models of fetal and neonatal alloimmune thrombocytopenia. Genes Dis. 2015; 2: 173-185.
- Kamphuis MM, Tiller H, van den Akker ES, et al. Fetal and Neonatal Alloimmune Thrombocytopenia: Management and Outcome of a Large International Retrospective Cohort. Fetal Diagn Ther. 2017; 41: 251-257.
- Curtis BR. Recent progress in understanding the pathogenesis of fetal and neonatal alloimmune thrombocytopenia. Br J Haematol. 2015; 171: 671-682.
- Xu XR, Gallant RC, Ni H. Platelets, immune-mediated thrombocytopenias, and fetal hemorrhage. Thromb Res. 2016; 141 Suppl 2: S76-79.
- Li C, Piran S, Chen P, et al. The maternal immune response to fetal platelet GPIbalpha causes frequent miscarriage in mice that can be prevented by intravenous IgG and anti-FcRn therapies. J Clin Invest. 2011; 121: 4537-4547.
- Tiller H, Kamphuis MM, Flodmark O, et al. Fetal intracranial haemorrhages caused by fetal and neonatal alloimmune thrombocytopenia: an observational cohort study of 43 cases from an international multicentre registry. BMJ Open. 2013; 3.
- Bussel JB, Zabusky MR, Berkowitz RL, et al. Fetal alloimmune thrombocytopenia. N Engl J Med. 1997; 337: 22-26.
- Peterson JA, McFarland JG, Curtis BR et al. Neonatal alloimmune thrombocytopenia: pathogenesis, diagnosis and management. Br J Haematol. 2013; 161: 3-14.
- Tiller H, Husebekk A, Ahlen MT, et al. Current perspectives on fetal and neonatal alloimmune thrombocytopenia - increasing clinical concerns and new treatment opportunities. Int J Womens Health. 2017; 9: 223-234.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002; 8: 1227-1234.
- Wang Y, Andrews M, Yang Y, et al. Platelets in thrombosis and hemostasis: old topic with new mechanisms. Cardiovasc Hematol Disord Drug Targets. 2012; 12: 126-132.
- Xu XR, Zhang D, Oswald BE, et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci. 2016; 53: 409-430.
- Zhu G, Zhang Q, Reddy EC, et al. The integrin PSI domain has an endogenous thiol isomerase function and is a novel target for antiplatelet therapy. Blood. 2017; 129: 1840-1854.
- Li J, van der Wal DE, Zhu G, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. 2015; 6: 7737.
- Ma L, Simpson E, Li J, et al. CD8+ T cells are predominantly protective and required for effective steroid therapy in murine models of immune thrombocytopenia. Blood. 2015; 126: 247-256.
- Li C, Li J, Li Y, et al. Crosstalk between Platelets and the Immune System: Old Systems with New Discoveries. Adv Hematol. 2012; 2012: 384685.
- Li C, Chen P, Vadasz B, et al. Co-stimulation with LPS or Poly I:C markedly enhances the anti-platelet immune response and severity of fetal and neonatal alloimmune thrombocytopenia. Thromb Haemost. 2013; 110: 1250-1258.
- Curtis BR. Genotyping for human platelet alloantigen polymorphisms: applications in the diagnosis of alloimmune platelet disorders. Semin Thromb Hemost. 2008; 34: 539-548.
- Sachs UJ. Fetal/neonatal alloimmune thrombocytopenia. Thromb Res. 2013; 131 Suppl 1: S42-46.
- Wu G, Zhou Y, Li L, et al. Platelet Immunology in China: Research and Clinical Applications. Transfus Med Rev. 2017; 31: 118-125.
- Chen P, Li C, Lang S, et al. Animal model of fetal and neonatal immune thrombocytopenia: role of neonatal Fc receptor in the pathogenesis and therapy. Blood. 2010; 116: 3660-3668.
- Ni H, Chen P, Spring CM, et al. A novel murine model of fetal and neonatal alloimmune thrombocytopenia: response to intravenous IgG therapy. Blood. 2006; 107: 2976-2983.
- Kovanlikaya A, Tiwari P, Bussel JB. Imaging and management of fetuses and neonates with alloimmune thrombocytopenia. Pediatr Blood Cancer. 2017; 64.
- Wang Y, Reheman A, Spring CM, et al. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest. 2014; 124: 4281-4293.
- Reheman A, Yang H, Zhu G, et al. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood. 2009; 113: 1809-1817.
- Yougbare I, Lang S, Yang H, et al. Maternal anti-platelet beta3 integrins impair angiogenesis and cause intracranial hemorrhage. J Clin Invest. 2015; 125: 1545-1556.
- Scheppke L, Murphy EA, Zarpellon A, et al. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood. 2012; 119: 2149-2158.
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005; 438: 954-959.
- Liu J, Fraser SD, Faloon PW, et al. A betaPix Pak2a signaling pathway regulates cerebral vascular stability in zebrafish. Proc Natl Acad Sci U S A. 2007; 104: 13990-13995.
- Eksteen M, Tiller H, Averina M, et al. Characterization of a human platelet antigen-1a-specific monoclonal antibody derived from a B cell from a woman alloimmunized in pregnancy. J Immunol. 2015; 194: 5751-5760.
- Santoso S, Wihadmadyatami H, Bakchoul T, et al. Antiendothelial alphavbeta3 Antibodies Are a Major Cause of Intracranial Bleeding in Fetal/Neonatal Alloimmune Thrombocytopenia. Arterioscler Thromb Vasc Biol. 2016; 36: 1517-1524.
- Winkelhorst D, Murphy MF, Greinacher A, et al. Antenatal management in fetal and neonatal alloimmune thrombocytopenia: a systematic review. Blood. 2017; 129: 1538-1547.
- Tiller H, Killie MK, Husebekk A, et al. Platelet antibodies and fetal growth: maternal antibodies against fetal platelet antigen 1a are strongly associated with reduced birthweight in boys. Acta Obstet Gynecol Scand. 2012; 91: 79-86.
- Kabir-Salmani M, Fukuda MN, Kanai-Azuma M, et al. The membrane-spanning domain of CD98 heavy chain promotes alpha(v)beta3 integrin signals in human extravillous trophoblasts. Mol Endocrinol. 2008; 22: 707-715.
- Snir A, Brenner B, Paz B, et al. Presence of Integrin alpha(IIb)beta 3 in early gestation human trophoblasts: possible involvement of fibrin as a matrix ligand. Thromb Res. 2010; 125: 253-256.
- Abdalvand A, Morton JS, Bourque SL, et al. Matrix metalloproteinase enhances big-endothelin-1 constriction in mesenteric vessels of pregnant rats with reduced uterine blood flow. Hypertension. 2013; 61: 488-493.
- Verlohren S, Geusens N, Morton J, et al. Inhibition of trophoblast-induced spiral artery remodeling reduces placental perfusion in rat pregnancy. Hypertension. 2010; 56: 304-310.
- Yougbare I, Zdravic D, Ni H. Angiogenesis and bleeding disorders in FNAIT. Oncotarget. 2015; 6; 15724-15725.
- Kumpel BM, Manoussaka MS. Placental immunology and maternal alloimmune responses. Vox Sang. 2012; 102: 2-12.
- Moffett A, Chazara O, Colucci F. Maternal allo-recognition of the fetus. Fertil Steril. 2017; 107: 1269-1272.
- Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. 2013; 31: 387-411.
- Croy BA, Luross JA, Guimond MJ, et al. Uterine natural killer cells: insights into lineage relationships and functions from studies of pregnancies in mutant and transgenic mice. Nat Immun. 1996; 15: 22-33.Croy BA, Luross JA, Guimond MJ, et al. Uterine natural killer cells: insights into lineage relationships and functions from studies of pregnancies in mutant and transgenic mice. Nat Immun. 1996; 15: 22-33.
- Vacca P, Montaldo E, Croxatto D, et al. Identification of diverse innate lymphoid cells in human decidua. Mucosal Immunol. 2015; 8: 254-264.
- Chakraborty D, Rumi MA, Konno T. et al. Natural killer cells direct hemochorial placentation by regulating hypoxia-inducible factor dependent trophoblast lineage decisions. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108: 16295-16300.
- Fu B, Li X, Sun R, et al. Natural killer cells promote immune tolerance by regulating inflammatory TH17 cells at the human maternal-fetal interface. Proc Natl Acad Sci U S A. 2013; 110: E231-240.
- Giuliani E, Parkin KL, Lessey BA, et al. Characterization of uterine NK cells in women with infertility or recurrent pregnancy loss and associated endometriosis. American journal of reproductive immunology. 2014; 72: 262-269.
- Carlino C, Stabile H, Morrone S, et al. Recruitment of circulating NK cells through decidual tissues: a possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood. 2008; 111: 3108-3115.
- Lash GE, Pitman H, Morgan HL, et al. Decidual macrophages: key regulators of vascular remodeling in human pregnancy. Journal of leukocyte biology. 2016; 100: 315-325.
- Althaus J, Weir EG, Askin F, et al. Chronic villitis in untreated neonatal alloimmune thrombocytopenia: an etiology for severe early intrauterine growth restriction and the effect of intravenous immunoglobulin therapy. American journal of obstetrics and gynecology. 2005; 193: 1100-1104.
- Eksteen M, Heide G, Tiller H, et al. Anti-human platelet antigen (HPA)-1a antibodies may affect trophoblast functions crucial for placental development: a laboratory study using an in vitro model. Reprod Biol Endocrinol. 2017; 15: 28.