
Diagnosis of ALCAPA in a 5-Year-Old Presenting with Atrial Arrhythmias
Pinting Chen MS1*, Anthony Khong MS2, Gary Ledley MD3, Vicki Mahan MD4
1,2Drexel University College of Medicine
3Hahnemann University Hospital, Drexel University College of Medicine
4St. Christopher’s Hospital for Children and Drexel University College of Medicine
Abstract
Anomalous left coronary artery to pulmonary artery (ALCAPA) is a congenital heart defect in which the left coronary artery (LCA) originates from the pulmonary trunk instead of from the aorta. This disease occurs in 1 in 300,000 births and, if untreated, 90% of these neonates die within the first year. Individuals who live beyond this tend to be asymptomatic and either experience sudden death at an average age of 35 or present with cardiac abnormalities, including myocardial ischemia, arrhythmia, or mitral regurgitation. There are various surgical interventions used to treat ALCAPA, and the establishment of a two coronary artery system is the preferred treatment of choice. We report a case of a five-year-old presenting with supraventricular tachycardia since birth who, upon diagnosis of ALCAPA by angiography, was treated by surgical ligation. Thirty years later, he returned to a cardiologist with symptoms of congestive heart failure. The initial presentation of this patient is unusual because the manifestation of ALCAPA usually occurs within the first few months of birth and because ligation is no longer the preferred method of intervention. We discuss this unique case, suggest possible associations between ALCAPA and arrhythmias, review the various surgical methods used to treat ALCAPA, and evaluate the long-term outcome of ligation.
Introduction
Anomalous left coronary artery to pulmonary artery (ALCAPA) is a cardiac anomaly in which the left coronary artery (LCA) arises from the pulmonary trunk instead of from the aorta (Figure 1). This heart defect occurs in 1 in 300,000 births. These neonates tend to present with non-specific symptoms of cardiopulmonary disease. These symptoms can include fussiness, cyanosis, discomfort, or problems with feeding, leading to an overall failure to thrive. Initial cardiac evaluation includes electrocardiography and echocardiography.
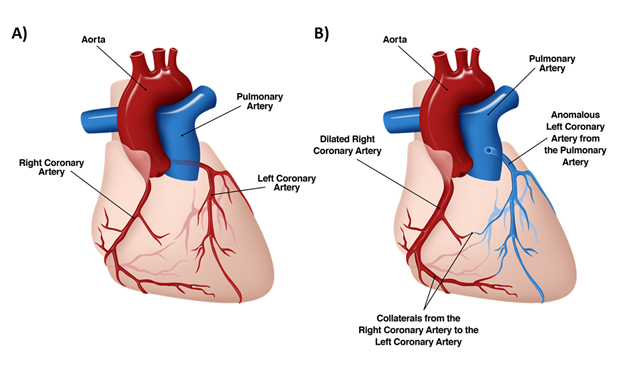
Figure 1: Anatomical diagram of the heart, frontal view. A, Normal heart with the LCA originating from the aorta. B, ALCAPA heart demonstrating extensive collateralizations and a dilated right coronary artery.
Ninety percent of patients born with ALCAPA and left untreated rarely live beyond their first year of life due to cardiac insufficiency, systemic ischemia, and myocardial infarction. In rare cases, however, ALCAPA patients who survive to adulthood are asymptomatic and usually experience sudden death at an average age of 351. In other cases, asymptomatic individuals can live much longer, to their seventies or eighties.
These individuals tend to present with a non-ALCAPA related complaint but upon further evaluation are found to have characteristic ALCAPA symptoms, murmurs (the most common), angina, dyspnea, palpitations, fatigue, syncope, and/or shortness of breath2,3. Diagnostic studies reveal arrhythmia, cardiac hypertrophy, retrograde flow from the left coronary artery to the pulmonary trunk (left-to-right shunt), a dilated right coronary artery (RCA), a left coronary artery (LCA) originating from the pulmonary artery, extensive collaterals from the RCA to the LCA, and/or mitral valve regurgitation – all characteristic of ALCAPA3–5. Numerous articles have attributed the longevity of adults with asymptomatic ALCAPA to the extensive intercoronary collaterals between the RCA and the LCA6–10.
There are various surgical interventions to repair ALCAPA and most are deemed successful with the individuals returning toward baseline post-surgery. Such surgical interventions include direct reimplantation of the LCA, the Takeuchi (transpulmonary baffling) repair, ligation of the LCA, the bay window technique and autologous pulmonary wall flap technique1,9,11,12. Patients who present with mitral regurgitation usually have improvement of their mitral regurgitation following surgery even though it is not a direct focus of the intervention11,13. While direct reimplantation of the LCA is now the preferred method, certain factors often contraindicate the use of this surgical intervention. Distance, location, and quality of the LCA are some of the primary factors, and surgeries are selected to avoid stretching, kinking, and to maintain the integrity of the artery1,11,14.
Case Report
We report a case of ALCAPA presenting as supraventricular tachycardia (SVT) unresponsive to medication in a 5-year-old male. Interestingly, though the patient records starting from 1981 state the patient had SVT, the electrocardiogram (EKG) done in 1983 when the patient was two years old shows accelerated junctional tachycardia (Figure 2). At four months of age, in 1981, the child was given 25 mcg of digoxin, given orally twice a day, to control his cardiac abnormality. This regimen was continued until 1983, when the dosage was doubled to 50 mcg. One year later, the dosage was again increased, this time to 75 mcg of digoxin, still given orally and twice a day. Compliance, however, was strongly in question as the patient often reported that he did not take his medications. During the first five years of life, the patient was hospitalized repeatedly for reported SVT. After each hospitalization, he was subsequently sent home with digoxin to control his arrhythmias. During one of his hospitalizations in 1985, he was given verapamil in the emergency room to control the tachycardia and digoxin was increased. Echocardiogram was not diagnostic and at 5 years old the patient was referred for diagnostic cardiac catheterization and electrophysiology. Studies revealed a dilated right coronary artery and collaterals stemming from the right coronary artery to supply the left coronary artery, which emptied into the main pulmonary artery. This confirmed a diagnosis of ALCAPA. Hemodynamic data reported a right ventricle oxygen saturation of 75% and 79-80% at the main pulmonary artery level. Right heart pressure and wedge pressure were normal while the left ventricular end-diastolic pressure was 12 mmHg. There was a 5 mm Hg gradient across the atrial septum and, post angiography, the left ventricular end-diastolic pressure was 18 mmHg (Figure 3). The findings of the electrophysiology studies showed refractory periods limited by atrial refractoriness and a retrograde Wenckebach phenomena across the AV node. Unsustained SVT was induced twice and demonstrated a cycle length of 305-310 msec, a duration of 9-15 beats, activation to be in the low septal right atrium to high right atrium, an Ae-H conduction of 210 msec, an H-Ae conduction of 100 msec, termination by block in the slow pathway, and a surface P wave up in lead 1 and down in lead AVF. A gap phenomena at the point where the A2-H2 interval was about to prolong was also encountered. These findings were thought to be consistent with SVT of the AV nodal re-entrant type, along with VA conduction with retrograde Wenckebach.
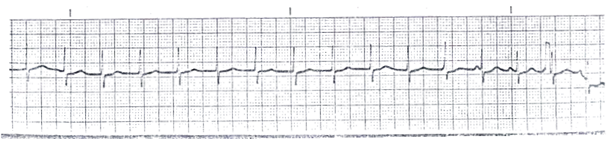
Figure 2: EKG, performed in 1983, showing an accelerated junctional rhythm in lead II.

Figure 3: Catheterization report with oximetry and pressure values. Step-up is seen from RV to MPA. On two different time periods, an increase in saturation was noted from the right ventricle at 74% to the branch pulmonary artery at 77% and from the right ventricle at 75% to the main pulmonary artery at 79-80%.
The patient was referred for surgery and ligation of the ALCAPA was performed in March of 1986. Multiple episodes of tachycardia, reported as SVT in the operative record, and severe hypotension occurred during the operation, causing the patient to require synchronized cardioversion and intravenous digoxin. Postoperative SVTs were treated with verapamil and vagal maneuvers. The patient was discharged on digoxin. He was followed by the pediatric cardiologist, and in 1994 a thallium perfusion scan reported findings compatible with an area of reversible ischemia involving the anterolateral wall of the left ventricular myocardium. The patient was then lost to follow-up at 12 years old. He later presented at 36 years old with symptoms of congestive heart failure and was seen by our adult cardiologist (GL). However, attempts to continue follow-up have proven unsuccessful.
Discussion
This is a rare presentation of a patient with ALCAPA as most patients present within the first 3 months of life when pulmonary vascular resistance decreases. Surgical ligation is no longer the preferred method of intervention. Atrial arrhythmias prompting further cardiac evaluation in patients with ALCAPA are unusual. Cardiac catheterization with electrophysiologic studies in this patient was done after multiple hospitalizations for “SVT.” Echocardiography was nondiagnostic and the cardiac catheterization finding of ALCAPA was not expected. This late presentation of ALCAPA and the survival of the patient to age five without treatment may be attributed to the extensive collateralization from the right coronary artery to the left coronary artery.
The surgical intervention, in this case, was ligation of the left coronary artery. At the time of surgery, reimplantation was felt to be contraindicated due to the positioning of the left coronary artery - the left coronary artery was found to originate from the posteromedial aspect of the pulmonary artery adjacent to the origin of the aorta. Additionally, the branching arteries of the left coronary artery, the LAD and circumflex arteries, were oriented in such a way that manipulation of these arteries could have resulted in kinking of the vessels. The surgeons, therefore, opted for ligation of the LCA to circumvent these problems and to avoid any additional complications. As mentioned above, though surgical ligation was once the primary form of treatment for ALCAPA, it is no longer the treatment of choice. Newer techniques have been developed for the described surgical problem and this method has now been replaced by other, more effective treatments that re-establish a two-coronary artery system. In fact, numerous studies have suggested that the late mortality rate of patients who undergo ligation is 25%15–17. Long-term surgical outcomes have improved with the use of new methods that involve the translocation of the coronary artery with the re-establishment of a two-coronary artery system. This method is superior to ligation, which relies on only a single coronary artery to supply the entire heart. The increased coronary perfusion provided is thought to play a vital role in the survival of patients with ALCAPA.
Sudden death is a common occurrence for those patients who are asymptomatic and live beyond their first year of life18. In these initially asymptomatic patients, ventricular fibrillation can manifest as the first symptom of ALCAPA. For some, ventricular fibrillation is the likely cause of sudden cardiac arrest and subsequent death, for which ALCAPA can only be diagnosed post-mortem. As ventricular fibrillation can lead to cardiac arrest and sudden death in ALCAPA patients, the establishment of an association between arrhythmias and ALCAPA can allow for earlier detection of the disease in patients who present with arrhythmia. In previous cases, arrhythmias (more specifically ventricular fibrillation) associated with ALCAPA have been attributed to the myocardial ischemia resulting from the absence of adequate flow to the left coronary artery6,7,18,19. Similarly, the AVNRT detected in this patient is also likely to be associated with ALCAPA due to problems with perfusion, which can ultimately affect conduction of the heart. To further emphasize this point, in the few cases in which patients are successfully resuscitated after cardiac arrest, diagnosed with ALCAPA, and treated with correction surgery, follow-up is usually uneventful, and ventricular fibrillation is reversed15,18–22.
References
- Ling Y, Bhushan S, Fan Q, Tang M. Midterm outcome after surgical correction of anomalous left coronary artery from the pulmonary artery. J Cardiothorac Surg. 2016; 11(1): 137. doi:10.1186/s13019-016-0535-7.
- Fierens C. A 72 year old woman with ALCAPA. Heart. 2000; 83(1): 2e-2. doi:10.1136/heart.83.1.e2.
- Gangadharan V, Sivagnanam K, Murtaza G, et al. Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery. J Investig Med High Impact Case Reports. 2017; 5(1): 232470961668462. doi:10.1177/2324709616684629.
- Castaldi B, Vida V, Reffo E, et al. Speckle Tracking in ALCAPA Patients After Surgical Repair as Predictor of Residual Coronary Disease. Pediatr Cardiol. 2017; 38(4): 794-800. doi:10.1007/s00246-017-1583-z.
- Midgley FM, Watson DC, Scott LP, et al. Repair of anomalous origin of the left coronary artery in the infant and small child. J Am Coll Cardiol. 1984; 4(6): 1231-1234. doi:10.1016/S0735-1097(84)80142-4.
- Bafani E, Shukla AC, DiNardo JA. Unrecognized anomalous origin of the left coronary artery from the pulmonary artery as a cause of ventricular fibrillation after patent ductus arteriosus ligation in an infant. Anesth Analg. 2007; 104(1): 81-83. doi:10.1213/01.ane.0000250365.25480.c5.
- Fudulu DP, Tulloh RMR, Wolf AR, et al. Anomalous Left Coronary From the Pulmonary Artery Presenting as Ventricular Fibrillation After Persistent Ductus Arteriosus Ligation. Ann Thorac Surg. 2015; 100(1): e9-e10. doi:10.1016/j.athoracsur.2015.04.015.
- Beitzke A, Luha O, Gamillscheg A. Atypical Bland-White-Garland syndrome with stenosis of the origin of the left coronary artery: Catheter intervention after mammary artery bypass stenosis and residual fistula to the pulmonary trunk. Pediatr Cardiol. 2000; 21(2): 161-164. doi:10.1007/s002469910028.
- Cohn LH. The Journal of Thoracic and Cardiovascular Surgery. 2012; 144(1): 4-6.
- Moodie DS, Cook SA, Gill CC, et al. Thallium-20 1 myocardial imaging in young adults with anomalous left coronary artery arising from the pulmonary artery. J Nucl Med. 1980; 21(11): 1076-1079.
- Roberts SM, Banbury T, Mehta A. A Rare Case of Anomalous Left Coronary Artery From the Pulmonary Artery (Bland-White-Garland Syndrome) in a 68-Year-Old Woman. Semin Cardiothorac Vasc Anesth. 2017; 21(2): 186-190. doi:10.1177/1089253216659146.
- Vilá Mollinedo LG, Jaime Uribe A, Aceves Chimal JL, Martínez-Rubio RP, Hernández-Romero KP. Case Report: ALCAPA syndrome: successful repair with an anatomical and physiological alternative surgical technique. F1000Research. 2016; 5(0): 1680. doi:10.12688/f1000research.8823.2.
- Birk E, Stamler A, Katz J, et al. Anomalous origin of the left coronary artery from the pulmonary artery: diagnosis and postoperative follow up. Isr Med Assoc J. 2000; 2(2): 111-114. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10804930%5Cnpapers2://publication/uuid/E206FF2D-9A91-4EA8-B099-9AD828A99767.
- Tsutsumi K. Surgical repair of anomalous connection of the left coronary artery. SAGE Open Med Case Reports. 2014; 2: 2050313X1455011. doi:10.1177/2050313X14550118.
- Wesselhoeft H, Fawcett J, Johnson A. Anomalous Origin of the Left Coronary Artery from the Pulmonary Trunk. Circulation. 1968; 38: 403-425.
- Wilson C, Dlabal P, McGuire S. Surgical treatment of anomalous left coronary artery from pulmonary artery: follow-up in teenagers and adults. Am Heart J. 1979; 98(4): 440-446.
- Wollenek G, Domanig E, Salzer-Muhar U, et al. Anomalous origin of the left coronary artery: a review of surgical management in 13 patients. J Cardiothorac Surg. 1993; 34(5): 399-405.
- Germanakis I, Dittrich S, Schlensak C, et al. Ventricular Fibrillation in a Teenager as First Manifestation of Anomalous Origin of Left Coronary Artery from Pulmonary Artery. Hell J Cardiol. 2003; 44: 155-158. http://www.hellenicjcardiol.org/archive/full_text/2003/2/2003_2_155.pdf.
- Frapier JM, Leclercq F, Bodino M, et al. Malignant ventricular arrhythmias revealing anomalous origin of the left coronary artery from the pulmonary artery in two adults. Eur J Cardio-Thoracic Surg. 1999; 15(4): 539-541.
- Weiss C, Willems S, Hofmann T, et al. Resuscitation in ventricular fibrillation as the first manifestation of Bland-White-Garland syndrome in adulthood. 1997: 447-451.
- Kreutzer U, Krulls-Munch J, Angres M, et al. Successful resuscitation of a patient with ventricular fibrillation. Hell J Cardiol. 1998; 87(7): 560-565. doi:10.1007/s003920050215.
- Viskin S, Belhassen B, Paredes A, et al. Anomalous origin of the left coronary artery from the pulmonary artery?: Report of an adult with ventricular fibrillation as the presenting symptom The ligamentum arteriosum?: An unreported origin of peripheral emboli diagnosed by transesophageal echocard. 1984;(124).