
Congenital Insensitivity to Pain Syndrome with Anhidrosis. Review of Literature
Vugar Nabi Nabyev1, Hakan Seneran2, Mehmet Cemalettin Aksoy3*
1Acibadem Ankara Hospital, Department of Orthopedics, Ankara, Turkey
2Selcuk University Faculty of Medicine, Department of Orthopedics, Selcuklu/Konya, Turkey
3Hacettepe University, Faculty of Medicine, Department of Orthopedics, Ankara, Turkey
Abstract
The congenital insensitivity to pain with anhidrosis (CIPA) is a rare autosomal recessive disease caused by mutations in NTRK1 gene (neurotrophic tyrosine kinase receptor 1) located in chromosome 1q21-22, encoding the tyrosinase domain receptor high affinity nerve growth factor. It is characterized by anhidrosis, insensitivity to painful stimuli and mental retardation. Given their low prevalence and the few reported cases, it is important to know its main features to be considered in the differential diagnosis in pediatric practice. The therapeutic approach of CIPA remains unclear. The preventive approach remains the only possible treatment of CIPA. Early surgical treatment for long bone fractures to prevent pseudo arthrosis and to allow early weightbearing decreasing the risk of further osteopenia. The choice of appropriate antibiotics and surgical debridement in cases of infection might prevent further destruction of joints.
Introduction
Congenital insensitivity to pain and anhidrosis (CIPA), also known as hereditary sensory and autonomic neuropathy type IV, is an extremely rare syndrome1, 2. According to literature, the first step in the diagnosis of CIPA syndrome is consideration of the clinical presentation based on the combination of three basic signs: insensitivity to pain, anhidrosis, and mental retardation2,3. The first reference to a similar pathology was mentioned by Dearborn in the early 1900s2, and it was published in 1963 by Swanson4. Other possible signs may be associated: impaired temperature sensation5, mandibular osteolysis6, facial alterations7 growth disturbances8; heterotopic ossification9,repetitive soft tissue and osseous infections of hematogenous origin10,self-mutilating behavior8. This condition occurs with an incidence of 1 in 125 million newborns6.All clinical manifestation shown as Table 1.
Table 1: Table summarizing the common clinical manifestations
| Sensory Nervous System • Lip biting,corneal scarring, and autoamputation of digits • Temperature sensation (i.e. perception of hot and cold) • Hypoalgesia |
| Autonomic Nervous System • Anhidrosis • Hyperpyrexia |
| Cognition • Intellectual disability • Impulsivity, hyperactivity, reckless behaviors • Delayed puberity |
| Gastrointestinal • Uncoordinated swallowing • Recurrent misdirection • Nausea and vomiting |
| Repiratory • Repeated aspiration pneumonia • Severe hypoxemia |
| Cardiovascular • Cardiovascular lability • Postural hypotension • Blotchy erythema |
| Musculoskeletal • Delayed cutaneos healing • Periungual lesions • Early loss of teeth • Fractures caused by minor traumas • Delay in bone union • Staphylococcus aureus osteitis • Septic fistulated pseudarthrosis • Multiple dislocations • Neuroarthropathic complications |
The pathogenesis of CIPA is characterized by a genetic loss-of-function mutation of the NTKR1 gene (locus 1q 21- 22)11-13. Multiple new mutations have been progressively described12,14. NTRK1 mutations imply an alteration in TrkA, an NGF receptor. NGF is involved in surveillance of nociceptive sensory neurons and sympathetic autonomic neurons and collaborates in the activation and homeostasis of other cellular types so that a NTRK1 mutation will cause deficient development of15, 16 the afferent somatic sensory system for pain and temperature, located in the dorsal root ganglion sensory neurons and the autonomic sympathetic neuronal system, which implies loss of the innervation of eccrine sweat glands by sympathetic neurons17, the central nervous system18, the bidirectional communication between the immune system and the nervous system (NGF has a relevant role in the signal pathway of B lymphocytes through three processes: Trk A phosphorylation, cytoskeleton assemblage, and MAP kinase activation). The molecular alteration in the function of NGF in turn also alters the normal process of fracture consolidation19. Normal osteoblast/osteoprogenitor differentiation and proliferation are hindered, tending to result in fibroblast differentiation of multipotent stromal mesenchymal cells and periosteal cells. Bone metabolism is also affected by the lack of nociceptive fibers, present not only in the skin but also in the skeletal system20. Due to the trophic role that nociceptive fibers may play in the skeletal system, bone fractures are very common21.
Diagnostic Tests
Diagnosis is mainly clinical, plus some laboratory tests8. Symptoms necessary for diagnosis are insensitivity to pain, anhidrosis and intellectual disability. However, the severity of these symptoms is highly variable. Skin biopsy may reveal a lack of eccrine sweat gland innervation. A biopsy of the sural nerve may reveal characteristic findings including reduced numbers of myelinated and unmyelinated small-diameter fibres with normal numbers of large diameter fibres22. An axonal flare test with a small amount of diluted histamine injected under the skin, fails to cause the normal flare around the site of injection8, 23. Molecular genetic testing can confirm the diagnosis but is not available on many occasions11.
Laboratory Findings
Plasma norepinephrine levels were very low or undetectable and failed to increase in the upright posture despite normal blood pressure24. Plasma epinephrine levels were normal and increased when patient was upright24. Plasma renin activity also increased appropriately with upright posture. Patients with pure autonomic failure also had very low levels of plasma norepinephrine both supine and when upright, but in contrast to patients with CIPA failed to maintain blood pressure upright4.
Neurological laboratory tests may provide additional information. Short-latency somatosensory evoked potentials show marked prolongation of the central conduction time25 and microneurography reveals abnormal activity of somatic A-delta and C fibers in the nerves of the skin26. A negative sympathetic skin response may also be helpful in the diagnosis due to the lack of sudomotor nerves in skin biopsy27,28.
Finally, molecular evaluation that reveals mutations of the NTKR1 gene provides a definitive diagnosis29.
Orthopedic Clinical Manifestations
Musculoskeletal manifestations are frequent in HSAN-IV , and include repeated fractures and joint dislocations (Figure 3 a,c), arthritis and osteomyelitis (Figure 3 c,d,e,f), avascular necrosis, and Charcot arthropathy(Figure 2 and 3 a,b), in both upper and the lower extremities30,31.
A review of literature with HSAN-IV showed that fractures are frequent between 1 and 7 years of age, here as other complications have no apparent age relationship32. Fractures and dislocations develop in HSAN-IV, even without any apparent trauma or following minor trauma, such as short falls8 (Fig.1 a,b).
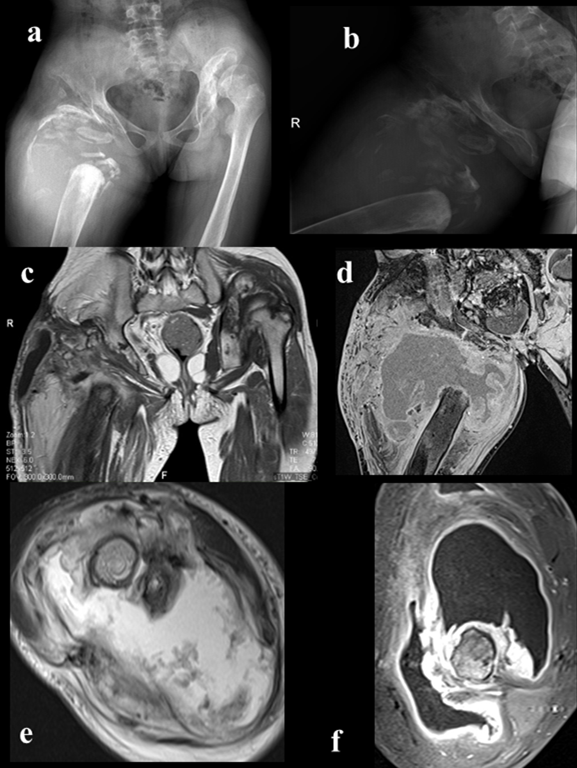
Figure 1: Right hip imaging;a,b)A-p and lateral view of right hip(Charcot’s joint, osteoarthritic neuropathy ),c,d)MRI imaging of process(frontal view),e,f)MRI imaging of affected hip(axial view)
Although the reason for these fractures and dislocations has not been clarified, decreased sensation, including deep sensation, intellectual disability, and mutilating behavior, may be related. Video gait analysis for patients with HSAN-IV has shown that younger patients walk faster with a longer stride length and higher heel contact angular velocity. than controls33. This finding may explain the high incidence of musculoskeletal trauma in the lower extremities. Charcot arthropathy may develop following malunion of fractures (Figure 4 c,e) avascular necrosis, and unreduced/recurrent dislocations in the absence of pain sensation.
Kawashima et al.34measured grip force and acceleration of a held object in patients with HSAN-IV. They found greater grip force during the object grasp-lift-holding task and greater fluctuation in acceleration of the object. Such impaired motor control ability may lead to overuse and/or misuse of the extremities and cause joint destruction (Figure 2 a,b,f).
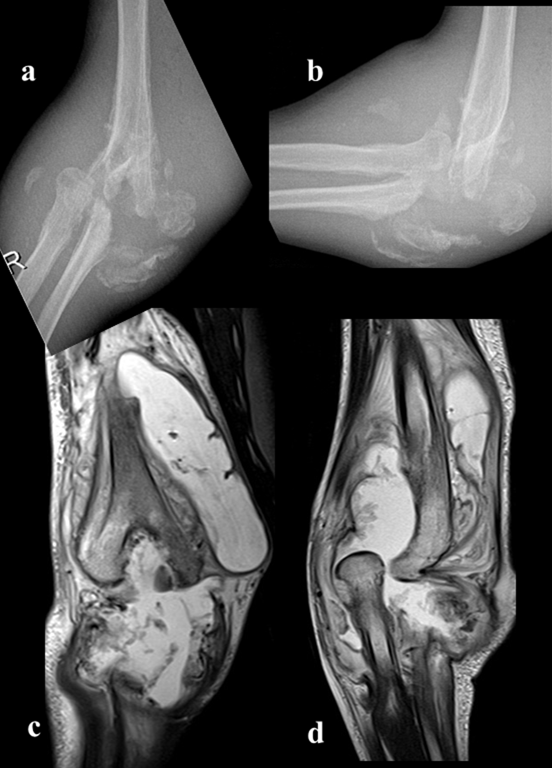
Figure 2: Right elbow disintegration due to septic course (Charcot’s joint, osteoarthritic neuropathy); a) right elbow A-P X-ray,b)right elbow lateral X-ray,c,d)right elbow MRG
The best therapeutic approach to patients with CIPA appears to be based on prophylactic measures such as braces for early weight bearing in nonsurgical fractures and accurate follow-up to avoid missing complications because of loss of pain sensation and intellectual disability. These may also lead to development of pressure sores. Even after surgical treatments, some patients cannot maintain non-weight bearing status and the fixation of fractures or reduction of dislocations can break down (Figure 3 a,b)8. Because surgical and conservative treatments have their drawbacks, prevention of trauma is important in HSAN-IV, although few comments on injury prevention have appeared in the literature.
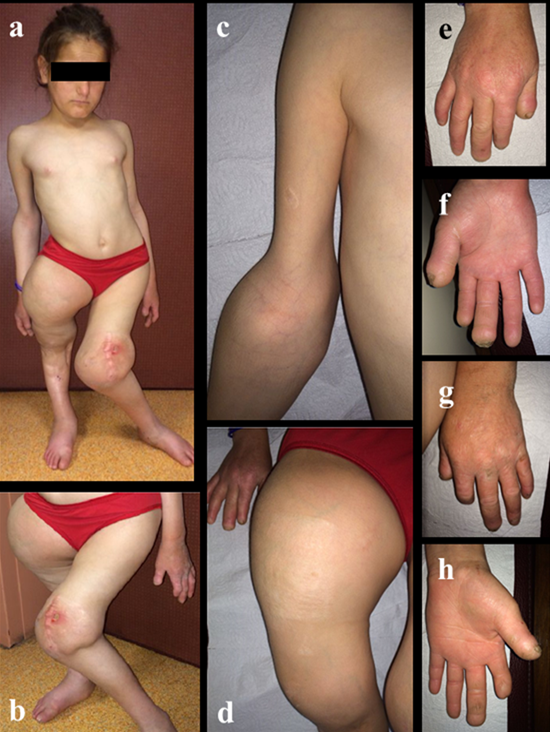
Figure 3: Clinical feature of patient suffered pain insensitivity syndrome; a)whole body is affected,b)left knee deformation due to septic arthritis,c)right elbow swelling and restriction of ROM,d)right hip swelling and deformation,e)right hand mutilation(dorsal view),f) right hand mutilation(palmar view),g)left hand mutilation(dorsal view),h) left hand mutilation(palmar view)
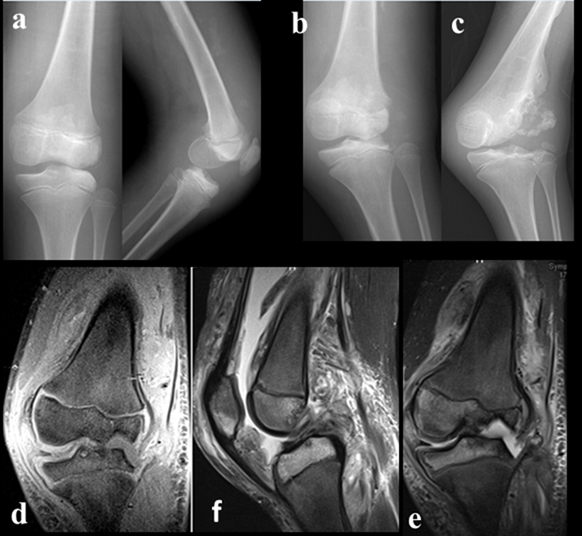
Figure 4: Affected left knee;a)initial A-p and lateral view of left knee when the patient first evaluated,b,d)after six month follow-up(x-ray and MRI),c,e)after year follow-up(x-ray and MRI)
The high incidence of infections in patients with CIPA is also problematic35. Skin and deep bone infections are the most common types, and Staphylococcus aureus is the most commonly involved pathogen36,37 (Figure 5). Resistance to antibiotics is a frequently occurring limitation in the treatment of these patients36.Temperature deregulation may cause recurrent fever, which may lead to death if not recognized early. Other complications such as trauma or soft tissue/bone infection may decrease condition of the survival rate, although all are treatable conditions if diagnosed in a timely manner38.
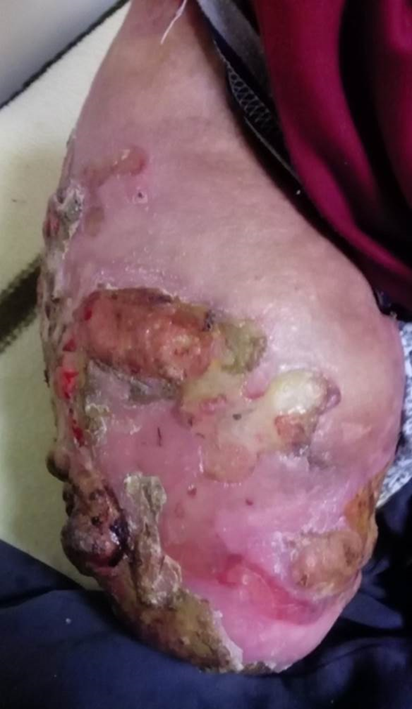
Figure 5: Affected extremities; Soft tissue infection after long transfemoral amputation
Wide surgical debridement and antibiotic therapy, corrective osteotomies, shoe raises or epiphysiodesis for shortening may be needed. These patients need regular follow-up with anticipation of complications and hospital re-admissions in the management of this rare syndrome8.
Discussion
Congenital insensitivity to pain with anhidrosis is caused by a mutation in the gene neurotrophic receptor tyrosinase 1 (NTRK1), which is located on chromosome 1q21-22, which encodes the tyrosinase domain of the receptor high affinity of nerve growth factor11,39. This gene alteration determines the failure of the differentiation and migration of cells the neural crest and, consequently, the absence complete of small myelinated fibers and not myelinated responsible for the perception of pain and temperature, as well as the absence of innervation of the sweat glands26. The prominent characteristics of the disease include fever due to anhidrosis, absence of sense of pain, painless ulcers in the structures inside the mouth and extremities, self-harm behavior, mild to severe mental retardation, myelination defect in the sural nerve biopsy and loss of small myelinized fibers25. Infection in the fingers and toes, lips and tongue and scarring are observed commonly6, 8. Keratoderma palmoplantaris is a typical finding in the advanced period6,40. Other problems include chronic bone and joint infections37. The first sign of this syndrome is fever secondary to anhidrosis, which has recurrent presentation since the period neonatal or from the first months of life. Due to the infrequency of this disorder and the absence of other signs that guide to its diagnosis, initially, they are described recurrent febrile episodes, such as fever of origin to be determined10. The febrile seizures occur frequently and described that 20% of the cases of death in the three first years of life are produced by hyperpyrexia18. As a consequence of the absence of painful and thermal sensitivity, are produced injuries at different levels. Common are alterations of the skin (lacerations, ulcers, infections, burns) and orthopedic (fractures multiple with hyperplastic bone formation, affecting femur and tibia; Charcot joints; limb dysmetria; scoliosis progressive osteomyelitis; septic arthritis; dislocations; amputations and self-mutilations32,41. The differential diagnosis among others hereditary neuropathies were mainly based at the age of onset of the clinical picture, in the marked insensitivity to pain and in the absence of sweating. Other important data for take into account for the differential diagnosis are the normal reflex response deep tendons and developmental delay maturity8. Clinical suspicion is confirmed with the result of skin biopsy. Malignancy was eventually ruled out based on histologic sampling. Inflammatory arthropathy is also a consideration, as in our case, when inflammatory changes predominate on imaging or there is polyarticular involvement31. An additional consideration is neuropathic joint related to chronic neuropathy caused by a long standing illness such as diabetes or vascular disease. Eventually, after excluding all other possibilities, the correct diagnosis of congenital insensitivity to pain was confirmed by gene testing36.
Conservative management can be useful in cases of mild deformities and disease consequences, thermoregulation is imperative in the management of CIPA and should include treatment of pyrexia with fanning, cooling blankets, cold drinks, paracetamol and/or non-steroidal anti-inflammatory drugs, and appropriate clothing advice42. Therapeutic approach with pamidronate as a compassionate use in this child obtained good results in preventing new fractures43.
Skeletal-system complications are more common in the lower limbs, almost all bones and joints may be affected. Szoke et al. reported that 58 % of patients with CIPA had osteoarticular disorders of the feet and ankles, 53 % of the knees, and 26 % of the hips31. Bar-On et al. reported that in 31 fractures in patients with congenital insensitivity to pain, 28 occurred in the lower limbs44. Fractures occurred in the early years of life, especially between the ages of 4 and 6 years. Fractures occur frequently at these ages because young children are very active. As children get older and become less active, the frequency of fractures decreases45. Furthermore, cardiovascular complications such as bradycardia, and hypotension following anesthesia, are common in patients with CIPA46,47.Therefore, surgical treatment of these patients may not always be advisable. Conservative therapy was used more frequently than surgery to manage fractures, On the other hand, use of casts, a representative conservative treatment, has drawbacks. First, the intellectual disability and loss of pain sensation of CIPA patients promotes instability. Second, there is a risk of developing pressure sores because of the sensory disturbance. Patients wore high-top sneakers and knee pads, and the parents covered the floor with sheets of soft material to prevent injuries. In addition, a few patients even used wheelchairs for locomotion to prevent accidental falls despite their walking ability. Some of published articles are described as Table 2.
Table 2: Table summarizing the published cases
| Published cases | Year of publication |
|---|---|
| Dearborn [2] | 1932 |
| Swanson [4] | 1963 |
| Tanaka et al. [15] | 1990 |
| Rosemberg et al. [5] rew | 1994 |
| Weier et al. [13] | 1995 |
| Indo et al. [11] | 1996 |
| Indo et al. [14] | 1997 |
| Grills and Schuijers [22] | 1998 |
| Miura et al. [13] | 2000 |
| Jarade et al. [9] review | 2002 |
| Bonkowsky et al. [39] | 2003 |
| Melamed et al. [19] | 2004 |
| Tunc¸bilek et al. [18] | 2005 |
| Schwarzkopf et al. [39] review | 2005 |
| Brandes and Stuth [44] | 2006 |
| Oliveira et al. [45] | 2009 |
| Lin et al. [12] | 2010 |
| Indo [15] | 2010 |
| Daneshjou et al. [6] | 2012 |
| Indo [26] | 2012 |
| Gao et al. [7] | 2013 |
| Fruchtman et al.[41] | 2013 |
| Yang et al. [45] | 2013 |
| Abdulla et al. [10] | 2014 |
| Pérez-López et al. [43] | 2015 |
| Nabiyev et al. [8] | 2016 |
Conclusion
HSAN are a very rare disorder with many diagnostic and treatment challenges. Data compilation of affected patients could provide a valuable resource of diagnosing and managing this disease.
A specialized and multidisciplinary approach is necessary to promote better development, reduce risks, treat complications and sequelae adequately. The participation and collaboration of parents and family members is fundamental, and they must be continuously trained to avoid all events that pose a possible risk to the patient. In the case presented, the dedication and affection of the family environment have been key in its favorable evolution.
References
- Gherlinzoni F, Gherlinzoni G. Neurogenic joint disease secondary to congenital insensitivity to pain. Italian journal of orthopaedics and traumatology. 1982; 8: 487-496.
- Ravichandra KS, Kandregula CR, Koya S, et al. Congenital Insensitivity to Pain and Anhydrosis: Diagnostic and Therapeutic Dilemmas revisited. International journal of clinical pediatric dentistry. 2015; 8: 75-81.
- Thrush DC. Congenital insensitivity to pain. A clinical, genetic and neurophysiological study of four children from the same family. Brain : a journal of neurology. 1973; 96: 369-386.
- Swanson AG. Congenital insensitivity to pain with anhydrosis. A unique syndrome in two male siblings. Archives of neurology. 1963; 8: 299-306.
- Rosemberg S, Marie SK, Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV). Pediatric neurology. 1994; 11: 50-56.
- Daneshjou K, Jafarieh H, Raaeskarami SR. Congenital Insensitivity to Pain and Anhydrosis (CIPA) Syndrome; A Report of 4 Cases. Iranian journal of pediatrics. 2012; 22: 412-416.
- Gao L, Guo H, Ye N, et al. Oral and craniofacial manifestations and two novel missense mutations of the NTRK1 gene identified in the patient with congenital insensitivity to pain with anhidrosis. PloS one. 2013; 8: e66863.
- Nabiyev V, Kara A, Aksoy MC. Multidisciplinary assessment of congenital insensitivity to pain syndrome. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2016; 32: 1741-1744.
- Jarade EF, El-Sheikh HF, Tabbara KF. Indolent corneal ulcers in a patient with congenital insensitivity to pain with anhidrosis: a case report and literature review. European journal of ophthalmology. 2002; 12: 60-65.
- Abdulla M, Khaled SS, Khaled YS, et al. Congenital insensitivity to pain in a child attending a paediatric fracture clinic. Journal of pediatric orthopedics Part B. 2014; 23: 406-410.
- Indo Y, Tsuruta M, Hayashida Y, et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature genetics. 1996; 13: 485-488.
- Lin YP, Su YN, Weng WC, et al. Novel neurotrophic tyrosine kinase receptor type 1 gene mutation associated with congenital insensitivity to pain with anhidrosis. Journal of child neurology. 2010; 25: 1548-1551.
- Mardy S, Miura Y, Endo F, et al. Congenital insensitivity to pain with anhidrosis (CIPA): effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Human molecular genetics. 2001; 10: 179-188.
- Weier HU, Rhein AP, Shadravan F, et al. Rapid physical mapping of the human trk protooncogene (NTRK1) to human chromosome 1q21-q22 by P1 clone selection, fluorescence in situ hybridization (FISH), and computer-assisted microscopy. Genomics. 1995; 26: 390-393.
- Indo Y. Nerve growth factor, pain, itch and inflammation: lessons from congenital insensitivity to pain with anhidrosis. Expert review of neurotherapeutics. 2010; 10: 1707-1724.
- Tanaka M, Sotomatsu A, Kanai H, et al. [Iron-dependent cytotoxic effects of dopa on cultured neurons of the dorsal root ganglia]. Rinsho shinkeigaku = Clinical neurology. 1990; 30: 379-383.
- Gargantilla P, Pintor E. [Congenital insensitivity to pain with anhidrosis in the literature]. Archivos argentinos de pediatria. 2015; 113: e197-198.
- Tuncbilek G, Oztekin C, Kayikcioglu A. Calcaneal ulcer in a child with congenital insensitivity to pain syndrome. Scandinavian journal of plastic and reconstructive surgery and hand surgery. 2005; 39: 180-183.
- Melamed I, Levy J, Parvari R, et al. A novel lymphocyte signaling defect: trk A mutation in the syndrome of congenital insensitivity to pain and anhidrosis (CIPA). Journal of clinical immunology. 2004; 24: 441-448.
- Derwin KA, Glover RA, Wojtys EM. Nociceptive role of substance-P in the knee joint of a patient with congenital insensitivity to pain. Journal of pediatric orthopedics. 1994; 14: 258-262.
- Hill EL, Elde R. Distribution of CGRP-, VIP-, D beta H-, SP-, and NPY-immunoreactive nerves in the periosteum of the rat. Cell and tissue research. 1991; 264: 469-480.
- Nolano M, Crisci C, Santoro L, et al. Absent innervation of skin and sweat glands in congenital insensitivity to pain with anhidrosis. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2000; 111: 1596-1601.
- Grills BL, Schuijers JA. Immunohistochemical localization of nerve growth factor in fractured and unfractured rat bone. Acta orthopaedica Scandinavica. 1998; 69: 415-419.
- Norcliffe-Kaufmann L, Katz SD, Axelrod F, et al. Norepinephrine deficiency with normal blood pressure control in congenital insensitivity to pain with anhidrosis. Annals of neurology. 2015; 77: 743-752.
- Shorer Z, Moses SW, Hershkovitz E, et al. Neurophysiologic studies in congenital insensitivity to pain with anhidrosis. Pediatric neurology. 2001; 25: 397-400.
- Indo Y. Nerve growth factor and the physiology of pain: lessons from congenital insensitivity to pain with anhidrosis. Clinical genetics. 2012; 82: 341-350.
- Hatzis J, Gourgiotou K, Koumelas D, et al. Congenital sensory neuropathy with anhidrosis (hereditary sensory neuropathy type IV). The Australasian journal of dermatology. 1992; 33: 103-107.
- Freeman R. Autonomic peripheral neuropathy. Lancet (London, England). 2005; 365: 1259-1270.
- Indo Y. Genetics of congenital insensitivity to pain with anhidrosis (CIPA) or hereditary sensory and autonomic neuropathy type IV. Clinical, biological and molecular aspects of mutations in TRKA(NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2002; 12 Suppl 1: I20-32.
- Minde J, Svensson O, Holmberg M, et al. Orthopedic aspects of familial insensitivity to pain due to a novel nerve growth factor beta mutation. Acta orthopaedica. 2006; 77: 198-202.
- Szoke G, Renyi-Vamos A, Bider MA. Osteoarticular manifestations of congenital insensitivity to pain with anhydrosis. International orthopaedics. 1996; 20: 107-110.
- Zhang Y, Haga N. Skeletal complications in congenital insensitivity to pain with anhidrosis: a case series of 14 patients and review of articles published in Japanese. Journal of orthopaedic science : official journal of the Japanese Orthopaedic Association. 2014; 19: 827-831.
- Zhang Y, Ogata N, Yozu A, et al. Two-dimensional video gait analyses in patients with congenital insensitivity to pain. Developmental neurorehabilitation. 2013; 16: 266-270.
- Kawashima N, Abe MO, Iwaya T, et al. Abnormal capacity for grip force control in patients with congenital insensitivity to pain. Experimental brain research. 2012; 218: 579-588.
- Karthikeyan M, Sreenivas T, Menon J, et al. Congenital insensitivity to pain and anhydrosis: a report of two cases. Journal of orthopaedic surgery (Hong Kong). 2013; 21: 125-128.
- Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: an update. Pain. 2003; 101: 213-219.
- Bronfen C, Bensahel H, Teule JG. [Orthopedic aspects of congenital insensitivity to pain]. Chirurgie pediatrique. 1985; 26: 193-196.
- Haga N, Kubota M, Miwa Z. Epidemiology of hereditary sensory and autonomic neuropathy type IV and V in Japan. American journal of medical genetics Part A. 2013; 161a: 871-874.
- Schwarzkopf R, Pinsk V, Weisel Y, et al. [Clinical and genetic aspects of congenital insensitivity to pain with anhidrosis]. Harefuah. 2005; 144: 433-437, 453, 452.
- Bonkowsky JL, Johnson J, Carey JC, et al. An infant with primary tooth loss and palmar hyperkeratosis: a novel mutation in the NTRK1 gene causing congenital insensitivity to pain with anhidrosis. Pediatrics. 2003; 112: e237-241.
- Fruchtman Y, Perry ZH, Levy J. Morbidity characteristics of patients with congenital insensitivity to pain with anhidrosis (CIPA). Journal of pediatric endocrinology & metabolism : JPEM. 2013; 26: 325-332.
- Zafeiriou DI, Vargiami E, Economou M, et al. Self-mutilation and mental retardation: clues to congenital insensitivity to pain with anhidrosis. The Journal of pediatrics. 2004; 144: 284.
- Perez-Lopez LM, Cabrera-Gonzalez M, Gutierrez-de la Iglesia D, et al. Update Review and Clinical Presentation in Congenital Insensitivity to Pain and Anhidrosis. Case reports in pediatrics. 2015; 2015: 589852.
- Bar-On E, Weigl D, Parvari R, et al. Congenital insensitivity to pain. Orthopaedic manifestations. The Journal of bone and joint surgery British volume. 2002; 84: 252-257.
- Yang L, Ji SF, Yue RJ, et al. Old fractures in two patients with congenital insensitivity to pain with anhidrosis: radiological findings. Clinical imaging. 2013; 37: 788-790.
- Brandes IF, Stuth EA. Use of BIS monitor in a child with congenital insensitivity to pain with anhidrosis. Paediatric anaesthesia. 2006; 16: 466-470.
- Oliveira CR, Paris VC, Pereira RA, et al. [Anesthesia in a patient with congenital insensitivity to pain and anhidrosis]. Revista brasileira de anestesiologia. 2009; 59: 602-609.